Advanced Asymmetric [3+2] Cyclization for High-Purity Chiral Carbocyclic Purine Nucleosides
Advanced Asymmetric [3+2] Cyclization for High-Purity Chiral Carbocyclic Purine Nucleosides
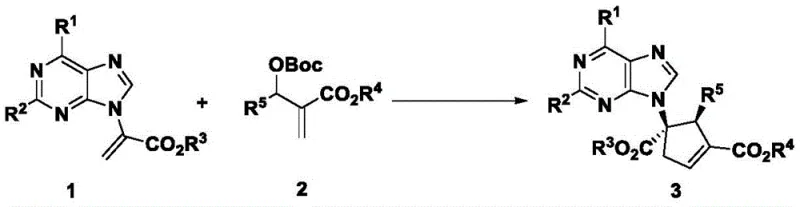
The synthesis of optically pure chiral five-membered carbocyclic purine nucleosides represents a critical challenge in modern medicinal chemistry, particularly given their pivotal role as active pharmaceutical ingredients (APIs) for treating viral infections such as HIV and HBV. Patent CN107698590B discloses a groundbreaking methodology that addresses these synthetic hurdles through an efficient asymmetric [3+2] cyclization reaction. This innovative process utilizes alpha-purine substituted acrylates and Morita-Baylis-Hillman (MBH) carbonates as primary building blocks, catalyzed by a specialized chiral SITCP monophosphine ligand. By bypassing the need for stoichiometric chiral auxiliaries, this technology offers a streamlined pathway to complex nucleoside analogs like Abacavir and Entecavir precursors, ensuring high diastereoselectivity and enantioselectivity while significantly simplifying the manufacturing workflow for global supply chains.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of chiral five-membered carbocyclic nucleosides has relied heavily on two primary strategies, both of which suffer from significant economic and operational inefficiencies. The first conventional route involves the meticulous design and multi-step synthesis of a chiral carbocyclic ring possessing the correct stereochemistry, followed by the coupling of this ring with a purine or pyrimidine base. This approach often necessitates the use of expensive chiral pool materials or resolution steps, leading to poor atom economy and substantial waste generation. Furthermore, the second traditional pathway requires the introduction of an amino group onto a pre-formed chiral ring to subsequently build the heterocyclic base, a process that is equally laborious and prone to low overall yields due to the cumulative losses across multiple synthetic transformations. These legacy methods impose severe bottlenecks on production scalability and cost-effectiveness.
The Novel Approach
In stark contrast, the methodology described in the patent introduces a direct, catalytic asymmetric [3+2] cyclization that fundamentally reshapes the synthetic landscape. By employing inexpensive, achiral starting materials such as alpha-purine substituted acrylates and MBH carbonates, the process eliminates the prerequisite for costly chiral substrates. The reaction is driven by a chiral organocatalyst that induces stereocontrol during the ring-forming event, directly assembling the complex five-membered carbocyclic core with high fidelity. This convergent strategy not only reduces the step count dramatically but also enhances the structural diversity accessible to chemists, allowing for the rapid generation of various analogs by simply modifying the substituents on the starting acrylates or carbonates, thereby accelerating lead optimization in drug discovery programs.
Mechanistic Insights into SITCP-Catalyzed Asymmetric Cyclization
The success of this transformation hinges on the precise interaction between the chiral SITCP catalyst and the reactive intermediates generated in situ. The mechanism likely proceeds through the formation of a zwitterionic species upon the nucleophilic attack of the phosphine catalyst on the MBH carbonate, generating an allylic phosphonium salt and a carbonate anion. This activated species then undergoes a [3+2] cycloaddition with the electron-deficient alpha-purine acrylate. The bulky chiral environment provided by the SITCP ligand, particularly the P6 variant featuring a binaphthyl backbone, effectively shields one face of the reacting species, enforcing a specific trajectory for the bond formation. This steric guidance is crucial for establishing the two contiguous stereocenters on the cyclopentene ring with high diastereo- and enantiocontrol, as evidenced by the experimental data showing dr values up to 10:1 and ee values reaching 96%.
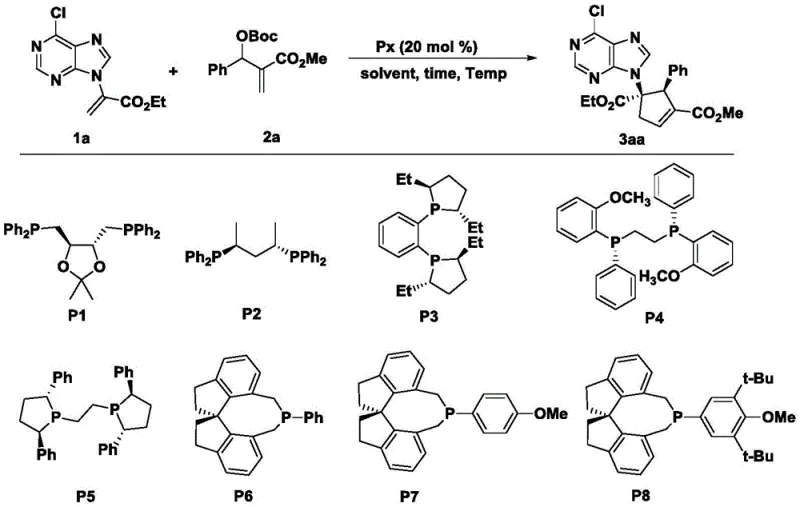
Furthermore, the robustness of this catalytic system allows for fine-tuning of the impurity profile through careful control of reaction parameters. The patent data highlights that maintaining the reaction temperature at -10°C is critical for maximizing enantioselectivity, as higher temperatures tend to erode the chiral induction provided by the catalyst. Additionally, the choice of solvent plays a non-trivial role; dichloromethane was identified as the optimal medium, balancing solubility and reaction rate without interfering with the catalyst's coordination sphere. By adhering to these strict parameters, manufacturers can ensure that side reactions, such as non-catalyzed background cyclization or polymerization of the acrylate, are minimized, resulting in a cleaner crude product that requires less intensive purification downstream.
How to Synthesize Chiral Carbocyclic Purine Nucleosides Efficiently
To implement this high-value synthesis in a laboratory or pilot plant setting, operators must follow a rigorous protocol that ensures the integrity of the moisture-sensitive catalyst and the stability of the reactive intermediates. The process begins with the preparation of the reaction vessel under an inert atmosphere, typically nitrogen, to prevent oxidation of the phosphine catalyst. The specific molar ratios of the alpha-purine acrylate, MBH carbonate, and the chiral SITCP catalyst (optimized at 20 mol%) must be precisely weighed and dissolved in anhydrous dichloromethane. The reaction mixture is then subjected to prolonged stirring at low temperatures (-10°C) for approximately four days to allow the slow but highly selective cyclization to reach completion. Detailed standard operating procedures for workup and purification are essential to maintain the high optical purity achieved during the reaction.
- Prepare the reaction mixture by combining alpha-purine substituted acrylate and MBH carbonate in dichloromethane under a nitrogen atmosphere.
- Add the chiral monophosphine catalyst (SITCP) at a loading of 20 mol% and maintain the reaction temperature at -10°C for approximately 4 days.
- Upon completion, quench the reaction, extract with dichloromethane and water, dry the organic phase, and purify the crude product via column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this asymmetric cyclization technology translates into tangible strategic benefits that extend beyond mere technical novelty. By shifting from multi-step chiral pool syntheses to a direct catalytic assembly, the process inherently reduces the number of unit operations required, which directly correlates to lower capital expenditure and reduced operational complexity. The reliance on achiral, commodity-grade starting materials mitigates the risk of supply disruptions often associated with specialized chiral building blocks, ensuring a more resilient and continuous supply of critical nucleoside intermediates for antiviral drug manufacturing.
- Cost Reduction in Manufacturing: The elimination of stoichiometric chiral reagents and the reduction in synthetic steps lead to a drastic simplification of the production process. Since the catalyst loading is relatively low (20 mol%) and the reaction proceeds with high yields (up to 93%), the overall cost of goods sold (COGS) is significantly optimized. Furthermore, the avoidance of expensive resolution steps or chiral chromatography for the starting materials removes a major cost driver typically found in traditional nucleoside synthesis, allowing for substantial margin improvement in the final API production.
- Enhanced Supply Chain Reliability: The use of robust, commercially available raw materials such as substituted acrylates and MBH carbonates ensures that the supply chain is not vulnerable to the bottlenecks of niche chiral suppliers. The reaction conditions are mild and do not require extreme pressures or hazardous reagents, facilitating easier transportation and storage of inputs. This stability allows for better inventory planning and reduces the lead time for high-purity pharmaceutical intermediates, enabling faster response to market demands for antiviral therapies.
- Scalability and Environmental Compliance: The process demonstrates excellent scalability potential, as evidenced by the successful execution of reactions on varying scales without loss of selectivity. The use of dichloromethane, while requiring proper handling, is a well-understood solvent in industrial settings with established recovery protocols. Moreover, the high atom economy of the [3+2] cyclization minimizes waste generation compared to linear synthetic routes, aligning with green chemistry principles and reducing the environmental burden associated with waste disposal and treatment.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and scope of this patented synthesis method. These insights are derived directly from the experimental data and optimization studies presented in the patent documentation, providing clarity on catalyst selection, substrate tolerance, and downstream processing capabilities for potential licensees and manufacturing partners.
Q: What are the key advantages of this asymmetric [3+2] cyclization method over traditional synthesis routes?
A: Unlike traditional methods that require expensive chiral starting materials and multi-step sequences, this patented approach utilizes readily available achiral raw materials and a single catalytic step to achieve high stereoselectivity (up to 96% ee) and excellent yields (up to 93%).
Q: Which catalyst system provides the optimal enantioselectivity for this transformation?
A: Screening data indicates that the chiral monophosphine ligand P6 (SITCP derivative) provides superior performance, achieving diastereomeric ratios of 9:1 and enantiomeric excesses exceeding 90% under optimized low-temperature conditions.
Q: Can the resulting nucleoside products be further functionalized for drug development?
A: Yes, the ester groups on the cyclopentene ring can be selectively reduced using reagents like NaBH4 or DIBAL-H to generate mono- or di-hydroxy derivatives, providing versatile intermediates for antiviral drug synthesis.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Chiral Carbocyclic Purine Nucleosides Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of this asymmetric [3+2] cyclization technology in advancing the production of next-generation antiviral agents. As a premier CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory discovery to industrial manufacturing is seamless and efficient. Our state-of-the-art facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications, including the precise determination of enantiomeric excess and diastereomeric ratios critical for regulatory compliance in the pharmaceutical sector.
We invite global pharmaceutical companies and research institutions to collaborate with us to leverage this cutting-edge synthesis route for your nucleoside pipeline. By partnering with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage you to contact us today to obtain specific COA data for our catalog of chiral intermediates and to discuss route feasibility assessments that can accelerate your project timelines and reduce overall development costs.