Advanced FeCl3-Catalyzed Synthesis of 5-Trifluoromethyl-1,2,4-Triazole Derivatives for Commercial Scale
Advanced FeCl3-Catalyzed Synthesis of 5-Trifluoromethyl-1,2,4-Triazole Derivatives for Commercial Scale
The pharmaceutical and agrochemical industries continuously demand efficient routes to nitrogen-containing heterocycles, particularly 1,2,4-triazole derivatives, which serve as critical scaffolds in numerous bioactive molecules. Patent CN111978265B discloses a groundbreaking preparation method for 5-trifluoromethyl substituted 1,2,4-triazole derivatives that addresses long-standing synthetic challenges. This technology leverages a ferric chloride-promoted cyclization strategy, utilizing inexpensive starting materials to achieve high yields under mild conditions. The significance of this innovation is underscored by the prevalence of the triazole motif in blockbuster drugs such as Maraviroc, Triazolam, Sitagliptin, and Deferasirox, as illustrated in the structural overview below.
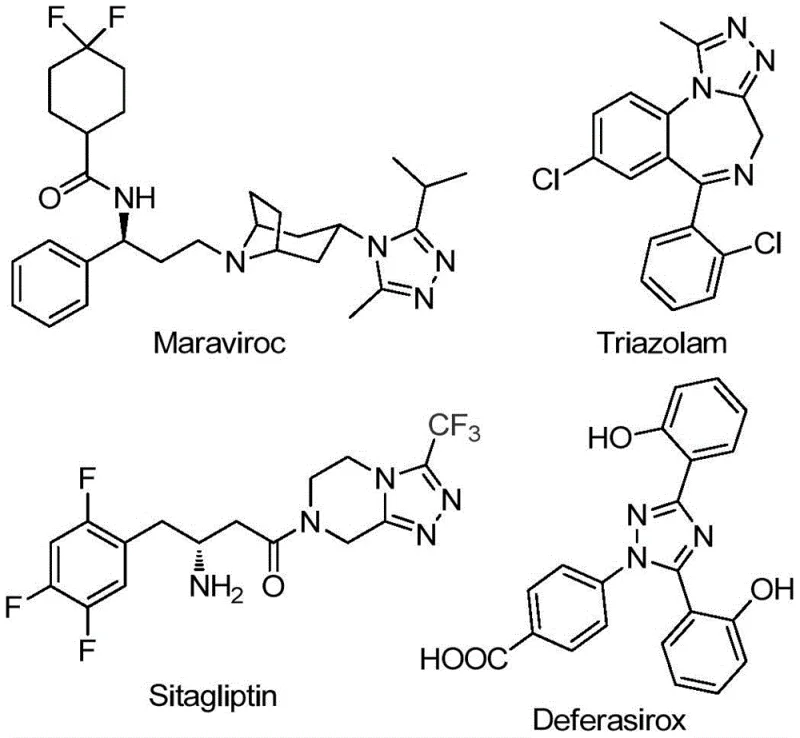
Furthermore, the introduction of trifluoromethyl groups into these heterocyclic systems significantly enhances electronegativity, metabolic stability, and lipophilicity, making these derivatives highly valuable for medicinal chemistry programs. The disclosed method represents a substantial leap forward in process chemistry, offering a reliable pathway for producing high-purity pharmaceutical intermediates without the logistical burdens associated with traditional protocols.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior to this invention, the synthesis of trifluoromethyl-substituted 1,2,4-triazoles was fraught with significant operational and economic inefficiencies. Literature reports typically describe five main approaches, including the condensation of 3,5-ditrifluoromethyl-1,3,4-oxadiazoles with primary amines or the cyclization of trifluoromethyl hydrazides with amidines. These conventional pathways are universally constrained by harsh reaction conditions that often require strict anhydrous and oxygen-free environments, drastically increasing operational costs and safety risks. Additionally, these methods frequently suffer from narrow substrate scopes; for instance, previous tandem cyclization reactions developed by the inventors themselves failed to accommodate alkyl hydrazones, thereby limiting the structural diversity of accessible 3-alkyl fluoro-1,2,4-triazoles. Such limitations hinder the rapid exploration of chemical space required for modern drug discovery.
The Novel Approach
In stark contrast, the novel approach detailed in patent CN111978265B utilizes a simple yet highly effective strategy involving cheap and readily available acyl hydrazides and trifluoroethylimidoyl chlorides. This method employs ferric chloride as a promoter, enabling the reaction to proceed efficiently without the need for complex inert atmosphere setups. The versatility of this new route is demonstrated by its ability to synthesize a wide array of derivatives, including those with alkyl, alkenyl, and various substituted aryl groups at the 3-position, as shown in the specific examples below. This expanded substrate tolerance allows chemists to access previously difficult-to-synthesize analogues, facilitating the optimization of lead compounds in pharmaceutical development pipelines.
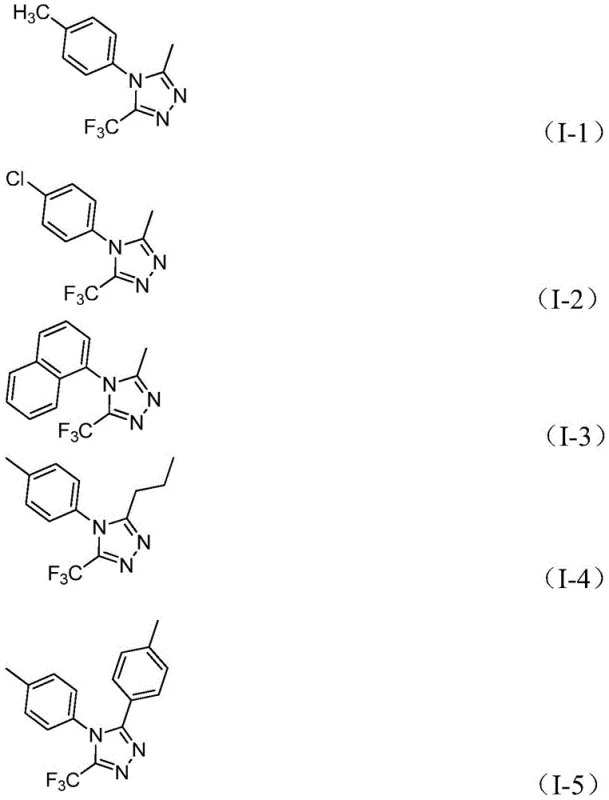
Mechanistic Insights into FeCl3-Catalyzed Cyclization
The core of this technological advancement lies in the dual-stage reaction mechanism that ensures high conversion and selectivity. The process initiates with a base-promoted intermolecular carbon-nitrogen bond formation between the trifluoroethylimidoyl chloride and the hydrazide, generating a trifluoroacetamidine derivative intermediate. This initial step is crucial for setting up the molecular architecture required for subsequent ring closure. Following this, the addition of the metal Lewis acid, specifically ferric chloride, triggers an intramolecular dehydration condensation. This Lewis acid promotion is pivotal, as it lowers the activation energy for the cyclization step, allowing the reaction to proceed at moderate temperatures while minimizing side reactions that could lead to impurity formation.
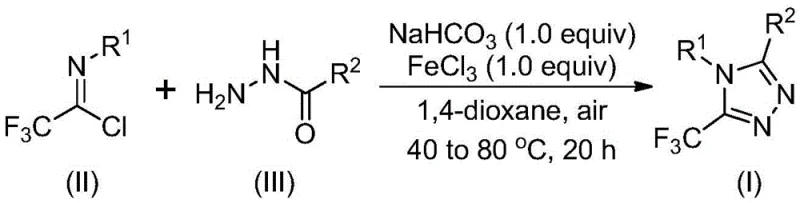
From an impurity control perspective, the use of sodium bicarbonate as a mild base in the first stage helps to neutralize generated acids without promoting excessive hydrolysis of the sensitive imidoyl chloride starting material. The sequential addition of reagents and the specific temperature profile—starting at 30-50°C and ramping to 70-90°C—further refine the reaction trajectory. This controlled thermal profile ensures that the intermediate forms completely before the cyclization begins, effectively suppressing the formation of polymeric byproducts or unreacted starting materials. For R&D directors, understanding this mechanistic nuance is vital for troubleshooting and optimizing the process for specific, complex substrates in custom synthesis projects.
How to Synthesize 5-Trifluoromethyl-1,2,4-Triazole Derivatives Efficiently
The synthesis protocol outlined in the patent provides a robust framework for laboratory and pilot-scale production. The procedure involves dissolving the reactants in a suitable aprotic solvent, with 1,4-dioxane identified as the optimal choice for maximizing conversion rates. The reaction is conducted in two distinct thermal phases to ensure complete transformation of the starting materials into the desired heterocyclic product. Detailed standard operating procedures regarding stoichiometry, mixing rates, and workup protocols are essential for maintaining batch-to-batch consistency. The following guide summarizes the critical steps derived from the patent data for implementing this methodology.
- Mix sodium bicarbonate, trifluoroethylimide chloride, and hydrazide in an organic solvent such as 1,4-dioxane at 30-50°C.
- Stir the mixture for 8 to 16 hours to allow base-promoted intermolecular carbon-nitrogen bond formation.
- Add ferric chloride catalyst and heat to 70-90°C for 6 to 10 hours to complete the cyclization, followed by purification.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the transition to this FeCl3-catalyzed methodology offers tangible strategic benefits beyond mere chemical efficiency. The primary advantage lies in the drastic simplification of the supply chain for raw materials. Unlike specialized reagents required for older methods, the starting materials here—acyl chlorides, hydrazine hydrate, and aromatic amines—are commodity chemicals widely available in the global market. This abundance mitigates the risk of supply disruptions and stabilizes pricing, ensuring a consistent flow of materials for continuous manufacturing operations. Furthermore, the elimination of expensive transition metal catalysts or exotic ligands reduces the direct material cost per kilogram of the final intermediate.
- Cost Reduction in Manufacturing: The economic impact of this process is driven by the replacement of costly and sensitive reagents with inexpensive alternatives like ferric chloride and sodium bicarbonate. By removing the requirement for rigorous anhydrous and oxygen-free conditions, the process significantly lowers infrastructure and utility costs associated with maintaining inert atmospheres. This simplification translates directly into reduced operational expenditure, allowing for more competitive pricing of the final active pharmaceutical ingredient intermediates without compromising on quality standards.
- Enhanced Supply Chain Reliability: The reliance on commercially available, off-the-shelf starting materials enhances the resilience of the supply chain. Since acyl chlorides and hydrazides are produced at scale by numerous chemical manufacturers globally, sourcing bottlenecks are minimized. This reliability is critical for long-term production contracts where continuity of supply is paramount. Additionally, the robustness of the reaction conditions means that production is less susceptible to delays caused by equipment failures related to specialized drying or degassing units.
- Scalability and Environmental Compliance: The patent explicitly notes that the method can be easily scaled up to the gram level and beyond, indicating strong potential for ton-scale commercial production. From an environmental standpoint, the use of iron-based catalysis is preferable to heavy metal alternatives, simplifying waste treatment and disposal protocols. The straightforward post-processing, involving filtration and standard column chromatography or crystallization, reduces the generation of complex hazardous waste streams, aligning with increasingly stringent global environmental regulations for chemical manufacturing.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These answers are derived directly from the experimental data and beneficial effects described in patent CN111978265B, providing clarity on the practical application of this method for industrial partners seeking to optimize their intermediate production capabilities.
Q: What are the key advantages of the FeCl3-catalyzed method over traditional synthesis?
A: The FeCl3-catalyzed method described in patent CN111978265B eliminates the need for harsh anhydrous or oxygen-free conditions required by conventional methods. It utilizes cheap, readily available starting materials like acyl hydrazides and tolerates a broader range of substrates, including alkyl hydrazones which previously failed to react.
Q: Is this synthesis method suitable for large-scale industrial production?
A: Yes, the process is highly scalable. The patent explicitly states that the method can be easily expanded to the gram level and beyond, providing convenience for industrial scale production applications due to its simple operation and lack of stringent environmental controls.
Q: What types of substituents are tolerated in this triazole synthesis?
A: The method demonstrates excellent functional group tolerance. R1 can be substituted or unsubstituted aryl groups with methyl, methoxy, halogen, or trifluoromethyl substituents. R2 allows for alkyl, alkenyl, or aryl groups, enabling the design of diverse 3,4-disubstituted triazole derivatives.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 5-Trifluoromethyl-1,2,4-Triazole Derivative Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that efficient synthetic methodologies play in accelerating drug development timelines. Our team of expert process chemists has extensively evaluated the FeCl3-catalyzed route described in CN111978265B and is fully prepared to deploy this technology for your specific project needs. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from benchtop discovery to full-scale manufacturing is seamless. Our facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications, guaranteeing that every batch of 5-trifluoromethyl-1,2,4-triazole derivative meets the highest industry standards.
We invite you to leverage our technical expertise to optimize your supply chain and reduce manufacturing costs. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and comprehensive route feasibility assessments to demonstrate how our advanced synthesis capabilities can support your long-term strategic goals in the pharmaceutical and agrochemical sectors.