Advanced Metal-Free Synthesis of Fluoroalkyl-Substituted Furans for Pharmaceutical Applications
Advanced Metal-Free Synthesis of Fluoroalkyl-Substituted Furans for Pharmaceutical Applications
The development of efficient synthetic routes for fluorinated heterocycles remains a paramount challenge in modern medicinal chemistry, particularly when constructing complex scaffolds like fluoroalkyl-substituted furans. Patent CN110357842B introduces a groundbreaking methodology that addresses these challenges by utilizing a selective four carbon-fluorine bond cleavage strategy. This innovation allows for the direct construction of fully substituted furan rings bearing both polyfluoroalkyl and sulfone moieties, structural motifs that are increasingly valued for their metabolic stability and lipophilicity in drug design. The significance of this technology lies not only in its chemical elegance but also in its potential to streamline the supply chain for high-value pharmaceutical intermediates. By leveraging readily available polyfluorinated peroxy compounds and organic sulfinates, this process bypasses the limitations of classical heterocycle synthesis, offering a robust platform for generating diverse chemical libraries.
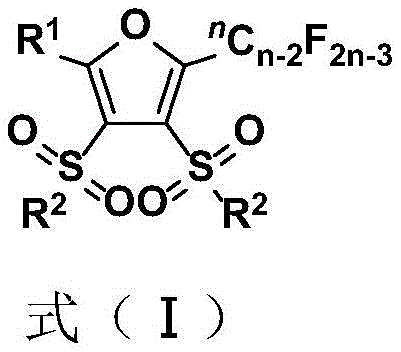
Furthermore, the ability to introduce fluorine atoms selectively into the furan skeleton opens new avenues for optimizing the physicochemical properties of bioactive molecules. Fluorine substitution is a well-established strategy in drug discovery to enhance binding affinity, metabolic stability, and membrane permeability. The compounds generated through this patented method exhibit a unique combination of a furan core, a polyfluoroalkyl chain, and sulfone groups, creating a privileged scaffold for targeting various biological receptors. For R&D directors seeking novel chemical space, this methodology provides access to structures that are difficult or impossible to obtain via conventional pathways, thereby accelerating the lead optimization phase in drug discovery programs.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of furan derivatives has relied heavily on classical methodologies such as the Feist-Benary synthesis, the Paal-Knorr synthesis, or transition-metal catalyzed rearrangements of alkene or alkyne derivatives. While these methods have served the chemical community well for decades, they suffer from significant drawbacks that hinder their application in modern, green manufacturing environments. The Feist-Benary and Paal-Knorr reactions often necessitate the use of strong inorganic acids or bases, which can lead to poor functional group compatibility and the degradation of sensitive substrates. Moreover, transition-metal catalyzed approaches, while powerful, introduce the risk of toxic metal contamination in the final product, a critical concern for pharmaceutical applications where strict limits on heavy metal residues are enforced by regulatory bodies.
Additionally, existing methods frequently struggle with the simultaneous introduction of fluorinated side chains during the ring construction process. Most traditional routes require multi-step sequences to install fluorine atoms post-cyclization, leading to lower overall yields and increased waste generation. The lack of atom economy and the generation of hazardous by-products in these conventional processes result in higher production costs and complex waste treatment requirements. For procurement and supply chain managers, these inefficiencies translate into longer lead times, higher raw material costs, and greater volatility in the supply of critical intermediates, making the search for alternative synthetic strategies an urgent priority.
The Novel Approach
In stark contrast to these legacy methods, the technology disclosed in Patent CN110357842B offers a streamlined, one-pot three-component coupling reaction that constructs the furan ring while simultaneously installing the fluoroalkyl and sulfone groups. This novel approach utilizes a base-promoted reaction between polyfluorinated peroxy compounds and organic sulfinates under mild thermal conditions. The elimination of transition metal catalysts is a game-changer, as it removes the need for expensive metal salts and the subsequent purification steps required to remove metal traces. This not only simplifies the workflow but also aligns with the principles of green chemistry by reducing the environmental footprint of the manufacturing process.
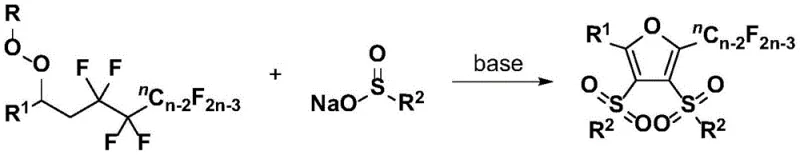
The versatility of this new method is evidenced by its broad substrate scope, accommodating a wide range of electronic and steric environments on both the peroxy and sulfinate components. Whether the starting materials contain electron-withdrawing groups like nitro or cyano, or electron-donating groups like methoxy or alkyl, the reaction proceeds with high efficiency and selectivity. This robustness ensures consistent product quality across different batches, a key metric for supply chain reliability. Furthermore, the reaction conditions are remarkably mild, typically operating between 50°C and 90°C in common solvents like tert-butanol or DMSO, which facilitates easy scale-up from laboratory benchtop to industrial reactor without the need for specialized high-pressure or cryogenic equipment.
Mechanistic Insights into Base-Promoted C-F Bond Cleavage and Cyclization
The core innovation of this synthesis lies in the unique activation and selective cleavage of four carbon-fluorine bonds within the polyfluoroalkyl peroxy precursor. Mechanistically, the reaction is initiated by the base promoter, which likely deprotonates the organic sulfinate or activates the peroxide species to generate reactive radical or anionic intermediates. These species then engage in a cascade of transformations that involve the fragmentation of the perfluoroalkyl chain. The selective breaking of C-F bonds is particularly noteworthy, as C-F bonds are among the strongest single bonds in organic chemistry, typically requiring harsh conditions or specialized reagents to cleave. The ability to achieve this under mild basic conditions suggests a highly tuned electronic interplay between the peroxide oxygen, the fluorine atoms, and the sulfinate sulfur.
Following the initial activation, the reaction proceeds through a cyclization event that constructs the five-membered furan ring. This step involves the coupling of two equivalents of the organic sulfinate with the activated fluoroalkyl fragment, effectively stitching together the carbon backbone of the heterocycle. The formation of the sulfone groups at the 3 and 4 positions of the furan ring occurs concurrently with the ring closure, demonstrating a high degree of convergency in the bond-forming events. This mechanistic pathway avoids the formation of unstable intermediates that often plague other fluorination strategies, thereby minimizing side reactions and maximizing the yield of the desired fully substituted furan product.
From an impurity control perspective, this mechanism offers distinct advantages. The absence of transition metals eliminates a major source of inorganic impurities, while the high selectivity of the C-F cleavage reduces the formation of defluorinated by-products or isomeric impurities. The reaction's tolerance to air atmosphere further indicates that the key intermediates are sufficiently stable to withstand ambient conditions, reducing the operational complexity associated with inert gas handling. For quality control teams, this translates to a cleaner crude reaction profile, which simplifies the downstream purification process and ensures that the final API intermediate meets stringent purity specifications with minimal effort.
How to Synthesize Fluoroalkyl-Substituted Furan Efficiently
Implementing this synthesis in a practical setting requires careful attention to reagent stoichiometry and reaction parameters to maximize yield and purity. The standard protocol involves mixing the polyfluorinated peroxy compound, the organic sulfinate, and a base promoter in a polar aprotic or protic solvent. The molar ratios are critical, with an excess of sulfinate and base typically employed to drive the reaction to completion and neutralize acidic by-products. The reaction is then heated to a moderate temperature, typically around 50°C to 90°C, and stirred for a period ranging from 5 to 24 hours depending on the specific substrate reactivity. Monitoring the reaction progress via thin-layer chromatography (TLC) is recommended to determine the optimal endpoint.
- Mix polyfluorinated peroxy compound, organic sulfinate, and alkali promoter in a suitable solvent such as tert-butanol or DMSO.
- Stir the reaction mixture in an air atmosphere at temperatures between 50°C and 90°C for 5 to 24 hours, monitoring progress via TLC.
- Upon completion, wash the product with water, extract with ethyl acetate, dry over anhydrous sodium sulfate, and purify via column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this synthetic route offers compelling economic and logistical benefits that extend beyond simple yield improvements. The primary driver of cost reduction is the elimination of transition metal catalysts. In traditional methods, the cost of palladium, copper, or other precious metal catalysts, combined with the expensive ligands often required, can constitute a significant portion of the raw material budget. By replacing these with inexpensive inorganic bases like cesium carbonate or potassium carbonate, the direct material cost is drastically reduced. Furthermore, the removal of metal catalysts obviates the need for specialized scavenging resins or complex extraction protocols to meet residual metal limits, simplifying the purification workflow and reducing solvent consumption.
- Cost Reduction in Manufacturing: The economic impact of this metal-free process is substantial when viewed through the lens of total manufacturing cost. Beyond the savings on catalysts, the mild reaction conditions (50-90°C) significantly lower energy consumption compared to high-temperature or high-pressure alternatives. The use of common, commodity-grade solvents like tert-butanol or DMSO, rather than exotic or highly regulated solvents, further contributes to cost efficiency. Additionally, the high atom economy of the three-component coupling means that a larger proportion of the starting mass ends up in the final product, reducing waste disposal costs. These factors combine to create a leaner, more cost-effective manufacturing process that enhances profit margins for the final API.
- Enhanced Supply Chain Reliability: Supply chain resilience is bolstered by the widespread availability of the key starting materials. Organic sulfinates and polyfluorinated peroxy compounds are commercially accessible from multiple global suppliers, reducing the risk of single-source dependency. The robustness of the reaction to air and moisture means that storage and handling requirements are less stringent, minimizing the risk of原料 degradation during transport or warehousing. This reliability ensures consistent production schedules and shorter lead times for customers, allowing pharmaceutical companies to maintain steady inventory levels of critical intermediates without the fear of unexpected supply disruptions caused by complex synthesis failures.
- Scalability and Environmental Compliance: Scaling this process from gram to kilogram or ton scale is straightforward due to the absence of hazardous reagents and extreme conditions. The reaction does not generate toxic heavy metal waste, simplifying compliance with increasingly strict environmental regulations regarding effluent discharge. The simplified workup procedure, involving standard aqueous washing and column chromatography, is easily adaptable to continuous flow chemistry or large batch reactors. This scalability ensures that the technology can meet the demands of commercial production without requiring significant capital investment in new infrastructure, making it an attractive option for contract development and manufacturing organizations (CDMOs) looking to expand their fluorinated intermediate portfolios.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this patented synthesis method. These answers are derived directly from the experimental data and beneficial effects described in the patent documentation, providing a clear understanding of the technology's capabilities and limitations. Understanding these details is crucial for technical teams evaluating the feasibility of integrating this route into their existing manufacturing pipelines.
Q: What are the primary advantages of this synthesis method over traditional Feist-Benary or Paal-Knorr reactions?
A: Unlike traditional methods that often require harsh acidic/basic conditions or toxic transition metal catalysts, this novel approach operates under mild conditions (50-90°C) using simple inorganic bases. It eliminates the need for expensive metal catalysts, thereby avoiding toxic metal residue issues and simplifying downstream purification, which is critical for pharmaceutical grade intermediates.
Q: Does this method allow for the introduction of diverse functional groups on the furan ring?
A: Yes, the method demonstrates excellent functional group tolerance and substrate universality. It successfully accommodates various substituents including halogens, nitro groups, cyano groups, and alkoxy groups on the aromatic rings of the starting materials, allowing for the synthesis of a wide library of structurally diverse fluoroalkyl-substituted furans.
Q: Is this process suitable for large-scale commercial production?
A: The process is highly amenable to scale-up due to its use of readily available starting materials like organic sulfinates and polyfluoroalkyl peroxy compounds. The reaction proceeds in common solvents like tert-butanol or DMSO under air atmosphere, removing the need for specialized inert gas equipment, which significantly lowers operational costs and complexity for industrial manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Fluoroalkyl-Substituted Furan Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of the synthetic methodology described in Patent CN110357842B for the production of high-value fluorinated intermediates. As a leading CDMO partner, we possess the technical expertise and infrastructure to rapidly translate this laboratory-scale innovation into commercial reality. Our team has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project can move seamlessly from clinical trial material to full-scale market supply. We are committed to maintaining stringent purity specifications and operating rigorous QC labs to guarantee that every batch of fluoroalkyl-substituted furan meets the highest industry standards.
We invite you to collaborate with us to leverage this advanced chemistry for your next drug development program. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality needs. Contact us today to request specific COA data for our catalog compounds or to discuss route feasibility assessments for your custom synthesis projects. Let us help you secure a reliable, cost-effective, and sustainable supply of these critical building blocks for the future of medicine.