Advanced Synthesis of Axial Chiral Indole-Naphthalene Compounds for Catalysis
Advanced Synthesis of Axial Chiral Indole-Naphthalene Compounds for Catalysis
The landscape of asymmetric catalysis is continuously evolving, driven by the demand for highly efficient, structurally diverse chiral ligands and organocatalysts. A significant breakthrough in this domain is detailed in patent CN110452150B, which discloses a novel preparation method for axial chiral indole-naphthalene compounds. These compounds, characterized by the general structure shown in Formula 9, represent a critical class of molecules capable of serving as potent organic micromolecular catalysts. The innovation lies in the ability to construct the complex axial chiral skeleton directly from racemic raw materials through a highly selective organocatalytic process. This approach not only simplifies the synthetic pathway but also ensures exceptional optical purity, addressing a long-standing challenge in the production of high-performance chiral catalysts for the pharmaceutical and fine chemical industries.
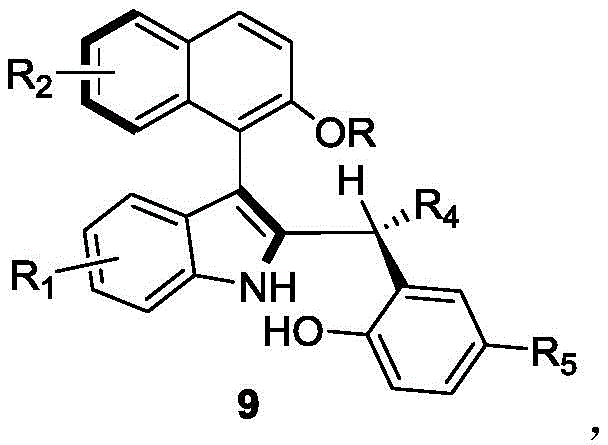
For research and development teams focusing on high-purity pharmaceutical intermediates, the structural versatility offered by this patent is paramount. The method accommodates a broad spectrum of substituents at various positions on the indole and naphthalene rings, including halogens, alkyl groups, and ester functionalities. This flexibility allows chemists to fine-tune the steric and electronic properties of the final catalyst, optimizing it for specific asymmetric transformations. By providing a robust route to these privileged scaffolds, the technology enables the rapid exploration of new catalytic systems without the prohibitive costs and low yields associated with classical resolution methods.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of axial chiral biaryl compounds, particularly those incorporating indole and naphthalene moieties, has relied heavily on transition-metal-catalyzed cross-coupling reactions or direct functionalization strategies. These conventional pathways often suffer from significant drawbacks that hinder their industrial applicability. Firstly, they typically require expensive and toxic transition metal catalysts, necessitating rigorous and costly purification steps to remove residual metals to meet pharmaceutical standards. Secondly, many existing methods struggle with substrate scope, failing to accommodate diverse functional groups without compromising yield or stereoselectivity. Furthermore, traditional approaches often involve multi-step sequences to install the chiral axis, leading to poor atom economy and increased waste generation, which contradicts the principles of green chemistry increasingly demanded by modern supply chains.
The Novel Approach
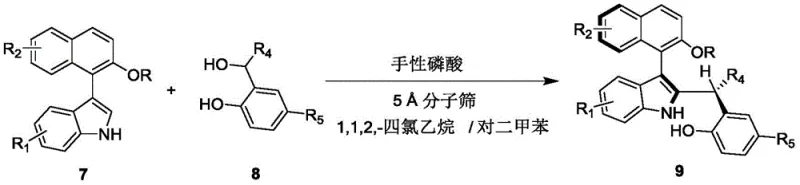
In stark contrast, the methodology described in the patent introduces a streamlined, one-step asymmetric addition reaction that constructs the axial chiral indole-naphthalene framework with remarkable efficiency. By employing a chiral phosphoric acid catalyst, the process achieves dynamic kinetic resolution of racemic starting materials (Formula 7 and Formula 8). This eliminates the need for pre-resolved enantiomers, effectively doubling the theoretical yield compared to classical kinetic resolution. The reaction proceeds under exceptionally mild conditions, typically at temperatures between 20°C and 30°C, using a mixed solvent system of 1,1,2,2-tetrachloroethane and p-xylene. This gentle environment preserves sensitive functional groups and minimizes energy consumption, making it an ideal candidate for cost reduction in chiral catalyst manufacturing.
Mechanistic Insights into Chiral Phosphoric Acid Catalyzed Asymmetric Addition
The success of this synthesis hinges on the precise mechanistic action of the chiral phosphoric acid catalyst, specifically derivatives based on binaphthyl, octahydrobinaphthyl, or spiro skeletons. The catalyst operates through a dual hydrogen-bonding activation mode. The acidic proton of the phosphoric acid activates the electrophilic species (likely the imine or similar intermediate derived from Formula 7), while the phosphoryl oxygen simultaneously coordinates with the nucleophile (Formula 8). This bifunctional activation brings the reactants into a rigid, well-defined chiral pocket created by the bulky substituents (such as the 9-anthracenyl groups in catalyst 6) on the catalyst backbone. This spatial confinement forces the reaction to proceed through a specific transition state, thereby exerting exquisite control over the formation of the new stereogenic axis and ensuring high enantioselectivity.
From an impurity control perspective, the mechanism offers distinct advantages. Because the reaction is highly selective and proceeds under mild thermal conditions, the formation of side products such as regioisomers or decomposition byproducts is significantly suppressed. The use of 5 Å molecular sieves as an additive further enhances the reaction efficiency by sequestering water, which could otherwise hydrolyze sensitive intermediates or deactivate the catalyst. This results in a cleaner crude reaction profile, simplifying downstream purification. For commercial scale-up of complex pharmaceutical intermediates, this means fewer chromatography cycles and higher overall recovery rates, directly translating to improved process economics and reduced environmental footprint.
How to Synthesize Axial Chiral Indole-Naphthalene Efficiently
The practical implementation of this synthesis is designed for operational simplicity, making it accessible for both laboratory-scale optimization and pilot plant production. The protocol involves mixing the indole-based substrate and the naphthol-derived nucleophile in the optimized solvent system, followed by the addition of the chiral catalyst and molecular sieves. The reaction is monitored via thin-layer chromatography (TLC) to ensure complete conversion before workup. Detailed standard operating procedures regarding stoichiometry, agitation speeds, and specific workup parameters are critical for maintaining the high stereochemical integrity observed in the patent examples.
- Prepare the reaction mixture by combining compound of formula 7 and compound of formula 8 in a mixed solvent of 1,1,2,2-tetrachloroethane and p-xylene.
- Add 5 Å molecular sieves and a chiral phosphoric acid catalyst (such as compound 6) to the mixture under stirring.
- Stir the reaction at 20-30°C until completion monitored by TLC, then filter, concentrate, and purify via silica gel column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this organocatalytic route presents a compelling value proposition centered on risk mitigation and cost efficiency. The shift from transition-metal catalysis to organocatalysis fundamentally alters the cost structure of the manufacturing process. By eliminating the need for precious metals like palladium or rhodium, the process removes a major source of raw material price volatility and supply chain fragility. Furthermore, the absence of heavy metals simplifies the regulatory compliance landscape, as there is no need for expensive metal scavenging resins or extensive testing for residual metal limits, which are stringent in API manufacturing.
- Cost Reduction in Manufacturing: The economic benefits of this process are driven by several key factors inherent to the reaction design. The use of racemic starting materials effectively halves the raw material cost compared to methods requiring enantiopure inputs. Additionally, the mild reaction temperature (25°C) drastically reduces energy consumption for heating or cooling, lowering the utility costs associated with large-scale production. The high yields and selectivity reported (often exceeding 90% yield and >95:5 dr) mean that less raw material is wasted on off-spec product, maximizing the throughput of the manufacturing facility and significantly reducing the cost per kilogram of the final active ingredient.
- Enhanced Supply Chain Reliability: The robustness of this synthetic route contributes to a more resilient supply chain. The starting materials (Formula 7 and 8) are structurally simple and can be sourced from multiple vendors or synthesized via established commodity chemical pathways, reducing the risk of single-source bottlenecks. The tolerance of the reaction to various functional groups implies that supply chain disruptions affecting specific substituted precursors can be mitigated by switching to alternative analogues without re-optimizing the entire process. This flexibility ensures reducing lead time for high-purity intermediates and maintains continuity of supply even in volatile market conditions.
- Scalability and Environmental Compliance: Scaling this process from gram to tonnage levels is facilitated by the benign reaction conditions. The absence of exothermic hazards associated with strong bases or reactive metal reagents enhances operational safety, a critical factor for large-scale chemical manufacturing. Moreover, the process aligns with green chemistry principles by utilizing organic small-molecule catalysts and generating less hazardous waste. This environmental compatibility simplifies waste disposal protocols and helps manufacturing partners meet increasingly strict environmental, social, and governance (ESG) targets, avoiding potential regulatory fines or shutdowns.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these axial chiral compounds. The answers are derived directly from the experimental data and technical specifications outlined in the patent documentation, providing a reliable basis for decision-making.
Q: What is the primary advantage of this synthesis method over traditional coupling reactions?
A: This method utilizes dynamic kinetic resolution of racemic raw materials via organocatalysis, allowing for the construction of the axial chiral skeleton in a single step under mild conditions (20-30°C), whereas traditional methods often require harsh coupling conditions and offer limited substrate scope.
Q: What level of enantioselectivity can be achieved with this process?
A: The process demonstrates excellent stereocontrol, achieving enantiomeric ratios (er) as high as 98:2 and diastereomeric ratios (dr) greater than 95:5 across a wide range of substrates, ensuring high optical purity suitable for catalytic applications.
Q: Can the resulting compounds be used directly as catalysts?
A: Yes, the synthesized axial chiral indole-naphthalene compounds serve as valuable precursors that can be derivatized into chiral phosphine catalysts, which have been proven effective in catalyzing asymmetric [4+1] cyclization reactions.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Axial Chiral Indole-Naphthalene Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of the technology described in CN110452150B for the next generation of asymmetric catalysts. As a premier CDMO partner, we possess the technical expertise to translate this innovative academic research into robust, GMP-compliant manufacturing processes. Our facilities are equipped to handle the specific solvent systems and purification requirements of this chemistry, ensuring that every batch meets the stringent purity specifications required for catalytic applications. We bring extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, guaranteeing that your supply needs are met with consistency and quality.
We invite you to collaborate with us to leverage this advanced synthesis for your specific projects. Our technical team is ready to provide a Customized Cost-Saving Analysis tailored to your volume requirements, demonstrating exactly how this organocatalytic route can optimize your budget. Please contact our technical procurement team today to request specific COA data, route feasibility assessments, and samples to evaluate the superior enantioselectivity of our axially chiral intermediates.