Advanced Metal-Free Synthesis of Trifluoromethyl 1,2,4-Triazines for Commercial Scale-Up
Advanced Metal-Free Synthesis of Trifluoromethyl 1,2,4-Triazines for Commercial Scale-Up
The rapid evolution of medicinal chemistry has placed trifluoromethyl-substituted heterocycles at the forefront of drug discovery, driven by their ability to enhance metabolic stability, lipophilicity, and bioavailability in lead compounds. Patent CN116253692A introduces a groundbreaking preparation method for trifluoromethyl substituted 1,2,4-triazine compounds, addressing critical bottlenecks in current synthetic methodologies. This innovation leverages a synergistic [3+3] cycloaddition strategy between chlorohydrazones and trifluoroacetyl sulfur ylides, facilitated by inexpensive potassium carbonate. The significance of this scaffold is underscored by its presence in potent bioactive molecules, ranging from PI3K inhibitors to dual c-Met/VEGFR-2 inhibitors, as illustrated in the structural diversity of modern therapeutics.
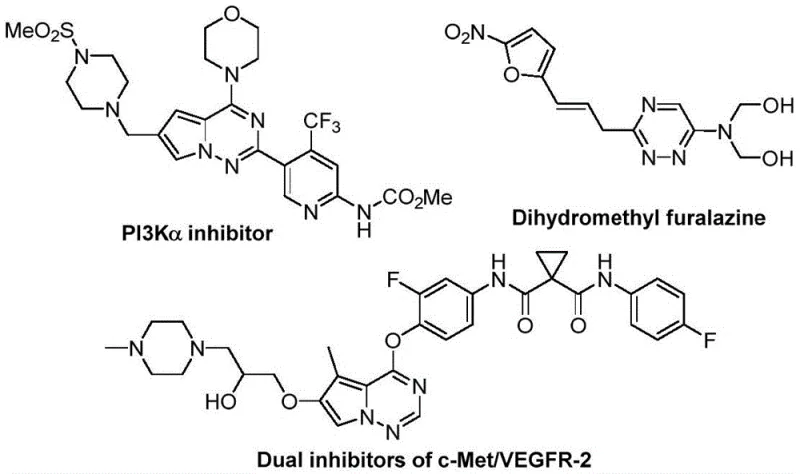
For R&D directors and process chemists, the introduction of this methodology represents a paradigm shift away from harsh, metal-dependent catalysis towards greener, more sustainable organic synthesis. The patent details a robust protocol that operates under ambient air conditions at temperatures between 20°C and 40°C, eliminating the stringent requirement for nitrogen protection often seen in sensitive heterocycle formations. By utilizing readily available starting materials and avoiding toxic heavy metals, this process not only streamlines the synthetic route but also aligns with the increasingly rigorous environmental, health, and safety (EHS) standards demanded by global regulatory bodies. The resulting 1,2,4-triazine derivatives exhibit high purity and structural integrity, making them ideal candidates for further functionalization in complex API synthesis pipelines.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of the 1,2,4-triazine core has relied heavily on the condensation of amidrazones with 1,2-diketones or alkynes, as well as multicomponent reactions involving hydrazides and dicarbonyl compounds. These traditional pathways are frequently plagued by significant inefficiencies, including the necessity for pre-synthesized, often unstable substrates that complicate supply chain logistics. Furthermore, conventional cyclization methods often suffer from poor atom economy and limited structural diversity, restricting the chemical space available for medicinal optimization. Many existing protocols require elevated temperatures, strong acids or bases, and transition metal catalysts, which introduce costly purification steps to remove trace metal residues—a critical concern for pharmaceutical intermediates intended for human consumption. The reliance on such苛刻 conditions often leads to lower yields and the formation of difficult-to-separate impurities, thereby inflating the overall cost of goods and extending development timelines.
The Novel Approach
In stark contrast, the novel approach disclosed in the patent utilizes a direct [3+3] cycloaddition between chlorohydrazones and trifluoroacetyl sulfur ylides, catalyzed by simple inorganic bases like potassium carbonate. This method bypasses the need for pre-functionalized diketones or complex multicomponent setups, instead relying on stable, commercially accessible precursors. The reaction proceeds smoothly in common organic solvents such as tetrahydrofuran (THF) at room temperature, demonstrating exceptional functional group tolerance. As shown in the general reaction scheme, the transformation is clean and efficient, generating dimethyl sulfoxide and hydrogen chloride as benign byproducts that are easily managed during workup. This streamlined process not only enhances reaction efficiency but also dramatically simplifies the downstream processing, allowing for the rapid generation of diverse trifluoromethyl-triazine libraries essential for high-throughput screening campaigns.
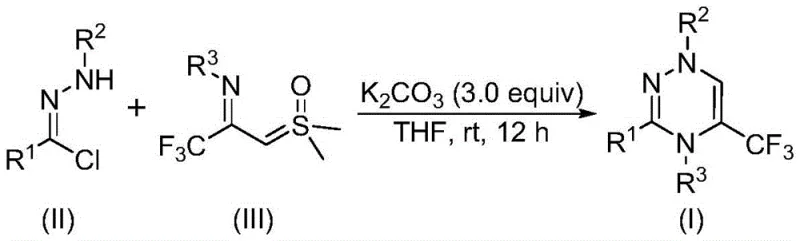
Mechanistic Insights into Potassium Carbonate-Promoted [3+3] Cycloaddition
The mechanistic elegance of this transformation lies in the in situ generation of a highly reactive nitrile imine intermediate. Under the promotion of potassium carbonate, the chlorohydrazone undergoes dehydrohalogenation to form the nitrile imine species, which serves as a potent 1,3-dipole. This intermediate then engages in a concerted [3+3] cycloaddition with the trifluoroacetyl sulfur ylide, a unique 1,3-dipolarophile characterized by its electron-deficient nature due to the trifluoromethyl group. The synergy between these two components facilitates the formation of the six-membered 1,2,4-triazine ring with high regioselectivity. The expulsion of dimethyl sulfoxide during the cyclization drives the reaction equilibrium forward, ensuring high conversion rates even under mild thermal conditions. This mechanism avoids the formation of radical species or high-energy transition states typically associated with metal-catalyzed C-H activation, thereby minimizing side reactions and preserving the integrity of sensitive functional groups on the aromatic rings.
From an impurity control perspective, the absence of transition metals eliminates the risk of metal-catalyzed decomposition or oligomerization, which are common pitfalls in heterocycle synthesis. The use of potassium carbonate, a mild and non-nucleophilic base, ensures that acid-sensitive groups remain intact while effectively scavenging the generated hydrogen chloride. The reaction profile suggests a clean conversion where the primary byproduct, dimethyl sulfoxide, is highly soluble in the reaction medium and easily removed during aqueous workup or chromatography. This high level of chemoselectivity is crucial for maintaining a narrow impurity profile, a key metric for R&D directors evaluating the feasibility of a route for GMP manufacturing. The ability to tune the electronic properties of the chlorohydrazone and sulfur ylide allows for precise control over the reaction kinetics, enabling the synthesis of complex derivatives without compromising yield or purity.
How to Synthesize Trifluoromethyl 1,2,4-Triazines Efficiently
The practical implementation of this synthesis is designed for ease of operation, requiring standard laboratory equipment and avoiding specialized high-pressure or cryogenic setups. The protocol involves mixing the chlorohydrazone, trifluoroacetyl sulfur ylide, and potassium carbonate in THF, followed by stirring at ambient temperature. This simplicity makes it highly attractive for both discovery chemistry and process development teams looking to rapidly access novel chemical space. The reaction time is typically between 10 to 14 hours, providing a convenient overnight workflow that fits seamlessly into standard operational schedules. Detailed standardized synthesis steps for scaling this reaction are provided in the guide below.
- Prepare the reaction mixture by adding potassium carbonate, chlorohydrazone, and trifluoroacetyl sulfur ylide into an organic solvent such as tetrahydrofuran (THF).
- Stir the reaction mixture at room temperature (20-40°C) in an air atmosphere for 10 to 14 hours to allow the [3+3] cycloaddition to proceed.
- Upon completion, filter the mixture, mix with silica gel, and purify via column chromatography to isolate the target trifluoromethyl substituted 1,2,4-triazine compound.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the economic implications of this patent are profound, primarily driven by the elimination of expensive and supply-constrained reagents. The substitution of precious metal catalysts with ubiquitous potassium carbonate represents a direct and significant reduction in raw material costs, while simultaneously removing the logistical burden associated with sourcing and handling sensitive organometallic reagents. Furthermore, the ability to run the reaction in air at room temperature drastically reduces energy consumption and infrastructure requirements, as there is no need for nitrogen generators, gloveboxes, or heated reactors. This operational simplicity translates into lower capital expenditure (CAPEX) for manufacturing facilities and reduced operational expenditure (OPEX) through decreased utility usage and simplified safety protocols. The robustness of the process ensures consistent batch-to-batch quality, mitigating the risk of production delays caused by failed reactions or complex purification challenges.
- Cost Reduction in Manufacturing: The complete removal of heavy metal catalysts from the synthetic route eliminates the need for costly metal scavenging resins and extensive analytical testing for residual metals, which are mandatory for pharmaceutical intermediates. By utilizing cheap, bulk-available inorganic salts and stable organic precursors, the overall cost of goods sold (COGS) is substantially lowered. The high atom economy of the [3+3] cycloaddition minimizes waste generation, further reducing disposal costs and enhancing the overall economic efficiency of the manufacturing process. This lean approach to synthesis allows for competitive pricing strategies in the global market for high-value heterocyclic building blocks.
- Enhanced Supply Chain Reliability: The starting materials, including chlorohydrazones and trifluoroacetyl sulfur ylides, are derived from commodity chemicals such as acyl chlorides, hydrazines, and trifluoroacetic acid, ensuring a stable and diversified supply base. Unlike specialized catalysts that may be sourced from single suppliers with long lead times, the reagents for this process are widely available from multiple global vendors, reducing the risk of supply chain disruptions. The mild reaction conditions also mean that the process can be easily transferred between different manufacturing sites without the need for specialized equipment validation, providing greater flexibility in production planning and inventory management.
- Scalability and Environmental Compliance: The process is inherently green, avoiding the use of toxic solvents or hazardous reagents, which simplifies regulatory compliance and environmental permitting. The scalability is demonstrated by the successful expansion to gram levels in the patent examples, with a clear path to kilogram and ton-scale production due to the exothermic nature being manageable at room temperature. The reduction in hazardous waste and the use of non-toxic promoters align with corporate sustainability goals, making this route preferable for companies aiming to reduce their carbon footprint. The straightforward workup involving filtration and chromatography ensures that the process remains efficient even at larger scales, facilitating rapid commercialization.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this trifluoromethyl triazine synthesis technology. These insights are derived directly from the experimental data and process descriptions within the patent documentation, providing clarity on substrate scope, reaction conditions, and scalability. Understanding these nuances is essential for technical teams evaluating the integration of this methodology into their existing R&D pipelines or manufacturing portfolios.
Q: What are the key advantages of this synthesis method over traditional routes?
A: This method eliminates the need for heavy metal catalysts and inert gas protection, operating efficiently at room temperature in air. It utilizes cheap, non-toxic potassium carbonate as a promoter, significantly simplifying post-treatment and reducing environmental impact compared to conventional condensation reactions.
Q: What is the substrate scope for this trifluoromethyl triazine synthesis?
A: The process demonstrates broad substrate tolerance, accommodating various substituted phenyl, naphthyl, and alkyl groups on the chlorohydrazone and sulfur ylide components. Functional groups such as methoxy, halogens (chloro, bromo), and trifluoromethyl are well-tolerated, yielding products with high structural diversity.
Q: Is this process suitable for large-scale industrial production?
A: Yes, the protocol is highly scalable due to its operational simplicity and safety profile. The use of inexpensive reagents, ambient reaction conditions, and straightforward purification via column chromatography makes it ideal for commercial scale-up from gram to multi-kilogram levels without complex engineering controls.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Trifluoromethyl 1,2,4-Triazine Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of this metal-free synthesis route for the production of high-value pharmaceutical intermediates. As a leading CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from benchtop discovery to full-scale manufacturing is seamless and efficient. Our state-of-the-art facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications, guaranteeing that every batch of trifluoromethyl 1,2,4-triazine delivered meets the highest industry standards. We are committed to leveraging our technical expertise to optimize this novel pathway, ensuring cost-effective and reliable supply for your critical drug development programs.
We invite you to collaborate with our technical procurement team to explore how this innovative synthesis can drive value for your organization. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into the economic benefits of switching to this metal-free protocol. We encourage you to contact us today to obtain specific COA data for our catalog compounds and to discuss route feasibility assessments tailored to your unique project requirements, ensuring a partnership built on transparency, quality, and shared success.