Revolutionizing Triazole Intermediate Production: Scalable Iodine-Catalyzed Synthesis for Global Pharmaceutical Supply Chains
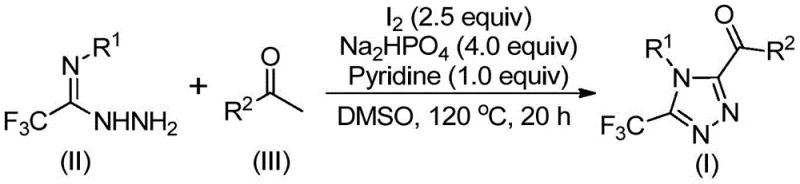
Patent CN113105402B introduces a groundbreaking methodology for synthesizing 3,4,5-trisubstituted 1,2,4-triazole compounds, representing a significant advancement in heterocyclic chemistry for pharmaceutical applications. This novel approach eliminates the need for anhydrous and oxygen-free conditions while avoiding toxic heavy metal catalysts, thereby addressing critical limitations in traditional triazole synthesis. The process utilizes readily available aryl ketones and trifluoroethylimide hydrazides as starting materials, enabling the construction of complex molecular architectures with trifluoromethyl and acyl functionalities that are essential for modern drug discovery. By operating under mild conditions with dimethyl sulfoxide as both solvent and reaction promoter, this method achieves remarkable substrate flexibility and functional group tolerance, making it particularly valuable for producing high-value pharmaceutical intermediates. The patent demonstrates successful gram-scale synthesis with yields ranging from 37% to 86%, establishing a robust foundation for commercial implementation in the fine chemical industry.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional approaches to synthesizing 3,4,5-trisubstituted 1,2,4-triazole compounds have been plagued by significant operational constraints and environmental concerns that hinder their industrial adoption. Conventional methods typically require stringent anhydrous and oxygen-free conditions that necessitate specialized equipment and increase production costs substantially, while the frequent use of transition metal catalysts introduces complex purification challenges and potential heavy metal contamination in the final product. These limitations become particularly problematic when attempting to incorporate both trifluoromethyl and acyl groups into the triazole scaffold, as existing methodologies often suffer from narrow substrate scope and poor functional group compatibility. Furthermore, the multi-step nature of traditional syntheses results in lower overall yields and increased waste generation, making them economically unviable for large-scale pharmaceutical manufacturing where purity specifications are exceptionally demanding. The requirement for expensive ligands and specialized reaction setups further compounds these issues, creating significant barriers to entry for manufacturers seeking to produce these valuable heterocyclic compounds at commercial scale.
The Novel Approach
The patented methodology presented in CN113105402B overcomes these longstanding challenges through an innovative iodine-promoted one-pot synthesis that operates under remarkably simple conditions. By leveraging the dual role of iodine as both oxidant and catalyst in dimethyl sulfoxide solvent, this process eliminates the need for expensive transition metals while maintaining excellent reaction efficiency across diverse substrate combinations. The method's brilliance lies in its sequential transformation where aryl ketones first undergo iodine/DMSO-mediated oxidation to form aryl diketones, which then react with trifluoroethylimide hydrazides through a dehydration-condensation-cyclization cascade to deliver the target triazoles in a single reaction vessel. Crucially, this approach functions effectively without anhydrous or oxygen-free requirements, dramatically simplifying process setup and reducing operational complexity while achieving yields between 37% and 86% across fifteen different substrate combinations. The elimination of heavy metal catalysts not only reduces purification costs but also ensures cleaner final products that meet the stringent quality standards required by pharmaceutical manufacturers, making this method uniquely positioned for commercial scale-up in the production of complex heterocyclic intermediates.
Mechanistic Insights into Iodine-Promoted Triazole Formation
The reaction mechanism begins with the iodine-mediated oxidation of aryl ketones in dimethyl sulfoxide to form α-iodo ketones, which subsequently undergo Kornblum oxidation to generate aryl diketone intermediates through a well-established pathway. These diketones then react with trifluoroethylimide hydrazides through a dehydration-condensation process to form hydrazone intermediates, which are critical precursors to the final triazole ring formation. The subsequent cyclization step is facilitated by the synergistic action of iodine and sodium dihydrogen phosphate in pyridine medium, where iodine acts as a Lewis acid to activate the carbonyl group while phosphate serves as a mild base to promote intramolecular nucleophilic attack. This cascade reaction proceeds through a six-membered transition state that enables efficient ring closure to form the 1,2,4-triazole core structure with simultaneous incorporation of both trifluoromethyl and acyl substituents at the 3 and 5 positions respectively. The mechanism's elegance lies in its ability to proceed under relatively mild conditions (110-130°C) without requiring inert atmosphere, making it exceptionally practical for industrial implementation while maintaining high regioselectivity throughout the transformation process.
Impurity control in this synthesis is achieved through multiple built-in mechanisms that ensure high product purity without extensive purification requirements. The reaction's inherent selectivity minimizes the formation of regioisomers by directing substitution specifically to the 3 and 5 positions of the triazole ring through electronic and steric control during the cyclization step. The use of dimethyl sulfoxide as both solvent and reaction promoter creates a homogeneous reaction environment that prevents side reactions commonly observed in heterogeneous systems, while the carefully optimized molar ratio of iodine (2.5 equiv), sodium dihydrogen phosphate (4.0 equiv), and pyridine (1.0 equiv) maintains precise control over reaction kinetics to avoid over-oxidation or decomposition pathways. Post-reaction purification is simplified through straightforward filtration followed by silica gel chromatography, with the patent demonstrating consistent production of high-purity compounds (as evidenced by NMR and HRMS data) that meet pharmaceutical intermediate standards without requiring additional purification steps that would increase production costs or reduce overall yield.

How to Synthesize Triazole Intermediates Efficiently
This patented methodology represents a significant advancement in triazole intermediate production, offering pharmaceutical manufacturers a practical solution to longstanding synthetic challenges. The process begins with simple aryl ketones and trifluoroethylimide hydrazides as starting materials, which undergo a carefully orchestrated sequence of oxidation, condensation, and cyclization steps under mild conditions that eliminate the need for specialized equipment or hazardous reagents. By operating at temperatures between 90-130°C in dimethyl sulfoxide solvent without requiring anhydrous or oxygen-free environments, this method dramatically simplifies process implementation while maintaining excellent yield and purity profiles across diverse substrate combinations. The following standardized synthesis protocol provides step-by-step guidance for manufacturing teams looking to implement this technology at scale.
- Combine aryl ketone with iodine in DMSO solvent at 90-110°C for 4-6 hours to form aryl diketone intermediates through iodine-mediated oxidation
- Add trifluoroethylimide hydrazide along with iodine (2.5 equiv), sodium dihydrogen phosphate (4.0 equiv), and pyridine (1.0 equiv) at 110-130°C for 12-20 hours to enable cyclization
- Perform post-reaction processing including filtration, silica gel mixing, and column chromatography purification to obtain high-purity triazole products
Commercial Advantages for Procurement and Supply Chain Teams
This innovative synthesis method delivers substantial value to procurement and supply chain decision-makers by addressing critical pain points in pharmaceutical intermediate manufacturing while enhancing overall operational efficiency. The elimination of expensive transition metal catalysts and specialized reaction conditions translates directly into reduced capital expenditure requirements and simplified facility setup, making this technology accessible to a broader range of manufacturing partners without significant infrastructure investment. Furthermore, the use of readily available starting materials from established chemical suppliers ensures consistent feedstock availability while minimizing supply chain vulnerabilities that often plague complex multi-step syntheses requiring rare or specialized reagents.
- Cost Reduction in Manufacturing: The complete avoidance of heavy metal catalysts eliminates both the initial procurement costs of expensive transition metals and the substantial downstream expenses associated with metal removal and purification processes required to meet pharmaceutical quality standards. This single modification creates significant cost savings throughout the production cycle while simultaneously reducing waste treatment costs associated with metal-contaminated byproducts. The simplified reaction setup also reduces energy consumption by eliminating the need for specialized inert atmosphere equipment and complex temperature control systems required by conventional methods.
- Enhanced Supply Chain Reliability: The reliance on commercially available aryl ketones and straightforward synthesis of trifluoroethylimide hydrazides from common precursors creates a robust supply chain foundation that minimizes vulnerability to single-source dependencies or geopolitical disruptions. This approach ensures consistent access to high-quality starting materials through multiple established chemical suppliers worldwide, while the method's tolerance for minor variations in raw material quality provides additional buffer against supply chain fluctuations that could otherwise disrupt production schedules.
- Scalability and Environmental Compliance: The demonstrated ability to scale from laboratory to commercial production (as evidenced by successful gram-scale reactions) combined with the absence of hazardous reagents makes this process inherently suitable for large-scale manufacturing without requiring significant process re-engineering. The elimination of toxic metal catalysts substantially reduces environmental impact while simplifying waste stream management, aligning with increasingly stringent global environmental regulations and corporate sustainability initiatives without compromising production efficiency or product quality.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial concerns regarding implementation of this patented triazole synthesis method in pharmaceutical manufacturing operations. These answers are derived directly from the experimental data and technical specifications provided in patent CN113105402B.
Q: Why does this method eliminate anhydrous/oxygen-free requirements?
A: The reaction utilizes dimethyl sulfoxide as both solvent and promoter in an iodine-mediated oxidation pathway that functions effectively under ambient atmospheric conditions without requiring inert gas protection or moisture control.
Q: How does avoiding heavy metal catalysts impact product purity?
A: Elimination of transition metals removes complex purification steps needed for metal residue removal, resulting in cleaner final products that meet stringent pharmaceutical quality specifications without additional processing.
Q: What substrate variations have been successfully demonstrated?
A: The method has been validated across fifteen different substrate combinations including various substituted phenyl groups with methyl, methoxy, chloro, fluoro substituents achieving yields from 37% to 86%.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Triazole Intermediate Supplier
Our patented methodology represents a transformative approach to triazole intermediate production that combines scientific innovation with practical manufacturing advantages for global pharmaceutical companies seeking reliable supply chain partners. NINGBO INNO PHARMCHEM brings extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production while maintaining stringent purity specifications through our state-of-the-art QC labs and rigorous analytical protocols. Our commitment to quality assurance ensures that every batch meets or exceeds pharmaceutical industry standards for purity and consistency, providing manufacturers with confidence in their critical raw material supply.
We invite procurement teams to initiate a Customized Cost-Saving Analysis to evaluate how this innovative synthesis method can optimize your specific manufacturing requirements. Contact our technical procurement team today to request detailed COA data and route feasibility assessments tailored to your production needs.