Scalable Iron-Catalyzed Synthesis of 2-Trifluoromethyl Quinazolinones for Advanced Drug Discovery
Scalable Iron-Catalyzed Synthesis of 2-Trifluoromethyl Quinazolinones for Advanced Drug Discovery
The landscape of pharmaceutical intermediate manufacturing is constantly evolving, driven by the need for more efficient, cost-effective, and environmentally sustainable synthetic routes. A significant breakthrough in this domain is detailed in patent CN111675662B, which discloses a novel preparation method for 2-trifluoromethyl substituted quinazolinone compounds. These heterocyclic scaffolds are of paramount importance in medicinal chemistry, serving as core structures for a vast array of bioactive molecules exhibiting anti-cancer, anticonvulsant, anti-inflammatory, and antifungal properties. The strategic introduction of a trifluoromethyl group into these frameworks further enhances their pharmacokinetic profiles by improving metabolic stability, lipophilicity, and bioavailability. This technical insight report analyzes the transformative potential of this iron-catalyzed methodology for global supply chains and R&D departments seeking reliable pharmaceutical intermediate suppliers.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of quinazolinones bearing trifluoromethyl functionalities has relied heavily on cyclization reactions involving synthons such as trifluoroacetic anhydride or ethyl trifluoroacetate reacting with substrates like anthranilamide, anthranilic acid, or isatoic anhydride. While these traditional pathways have served the industry for decades, they are increasingly recognized for their significant drawbacks in a modern commercial context. These methods often necessitate severe reaction conditions that can compromise sensitive functional groups, leading to complex impurity profiles that are difficult to purge. Furthermore, the reliance on expensive fluorinating agents and the frequent requirement for stoichiometric amounts of harsh reagents result in elevated production costs and substantial waste generation. The narrow substrate scope of many legacy protocols also limits the ability of medicinal chemists to rapidly explore structure-activity relationships (SAR), thereby slowing down the drug discovery pipeline.
The Novel Approach
In stark contrast to these legacy issues, the methodology outlined in CN111675662B introduces a streamlined and robust alternative utilizing readily available trifluoroethylimidoyl chloride and isatin derivatives as starting materials. This innovative route leverages a cheap metal iron catalyst to drive a series of cyclization reactions that efficiently construct the target quinazolinone core. As illustrated in the general reaction scheme below, this approach not only simplifies the synthetic sequence but also dramatically improves the overall atom economy.
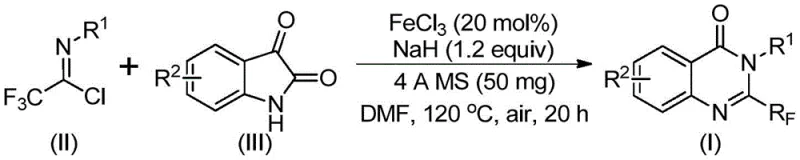
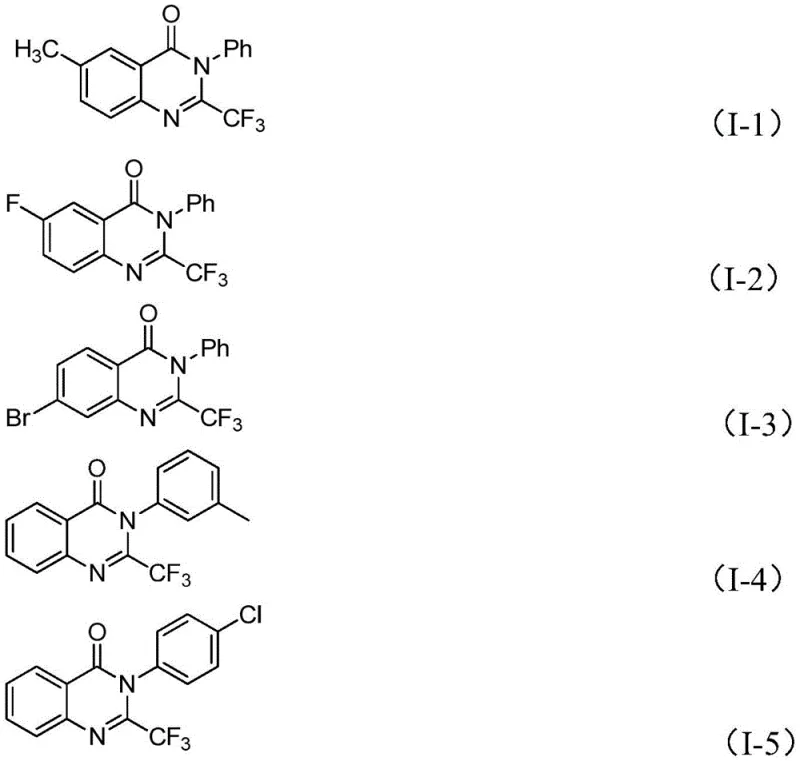
The versatility of this new method is evidenced by its broad functional group tolerance, allowing for the incorporation of diverse substituents such as methyl, fluoro, bromo, chloro, and nitro groups at various positions on the aromatic rings. Specific examples, including compounds (I-1) through (I-5) shown below, demonstrate the practical applicability of this chemistry in generating high-value intermediates with excellent yields.
Mechanistic Insights into FeCl3-Catalyzed Cyclization
At the heart of this synthetic advancement lies a sophisticated yet operationally simple catalytic cycle mediated by ferric chloride (FeCl3). The reaction mechanism initiates with the activation of the trifluoroethylimidoyl chloride by the Lewis acidic iron species, facilitating a nucleophilic attack by the nitrogen of the isatin derivative. This initial step forms a key trifluoroacetamidine intermediate, setting the stage for the subsequent ring closure. The presence of sodium hydride acts as a crucial base promoter, ensuring the deprotonation steps necessary for bond formation proceed smoothly. Following the initial coupling, the system undergoes an iron-catalyzed decarbonylation and cyclization sequence. This tandem process effectively excises the carbonyl oxygen from the isatin moiety and rearranges the molecular skeleton to form the stable quinazolinone ring system. The use of 4A molecular sieves in the reaction mixture plays a pivotal role in scavenging trace moisture, which could otherwise hydrolyze the sensitive imidoyl chloride or deactivate the catalyst, thereby ensuring high conversion rates and reproducibility.
From an impurity control perspective, this mechanism offers distinct advantages over acid-mediated cyclizations. The mild nature of the iron catalyst minimizes side reactions such as polymerization or over-fluorination, which are common pitfalls in trifluoromethylation chemistry. The specific interaction between the iron center and the nitrogen atoms likely stabilizes transition states that favor the desired regioisomer, resulting in a cleaner crude reaction profile. This inherent selectivity reduces the burden on downstream purification processes, a critical factor for procurement managers focused on cost reduction in API manufacturing. By understanding these mechanistic nuances, R&D teams can better optimize reaction parameters such as temperature gradients (starting at 40°C and ramping to 120°C) to maximize yield while minimizing energy consumption.
How to Synthesize 2-Trifluoromethyl Quinazolinones Efficiently
Implementing this synthesis in a laboratory or pilot plant setting requires adherence to specific procedural guidelines to ensure safety and optimal yield. The process involves a two-stage heating protocol where the reactants are first stirred at a moderate temperature to allow for initial adduct formation before being heated to promote the cyclization and decarbonylation steps. The use of polar aprotic solvents like DMF is preferred to ensure adequate solubility of the inorganic base and the organic substrates. Detailed standardized operating procedures regarding reagent addition rates, stirring speeds, and quenching protocols are essential for consistent results.
- Mix ferric chloride, sodium hydride, 4A molecular sieves, trifluoroethylimidoyl chloride, and isatin derivative in an organic solvent like DMF.
- Stir the reaction mixture at 40°C for 8-10 hours to initiate the coupling, then heat to 120°C for 18-20 hours to complete the cyclization.
- Filter the reaction mixture, purify via silica gel column chromatography to isolate the target 2-trifluoromethyl substituted quinazolinone compound.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the transition to this iron-catalyzed methodology represents a strategic opportunity to optimize sourcing strategies and reduce total landed costs. The shift away from precious metal catalysts or exotic fluorinating reagents towards commodity chemicals like ferric chloride and isatin significantly de-risks the supply chain. Since these raw materials are produced on a massive industrial scale globally, their availability is high, and price volatility is low compared to specialized organometallic complexes. This stability allows for more accurate long-term budgeting and contract negotiations with vendors.
- Cost Reduction in Manufacturing: The economic impact of replacing expensive catalysts with inexpensive iron salts cannot be overstated. Ferric chloride is a bulk chemical with a fraction of the cost of palladium or rhodium-based systems often used in cross-coupling reactions. Furthermore, the high atom efficiency of this cyclization means less raw material is wasted as byproducts. The simplified work-up procedure, which primarily involves filtration and standard chromatography, eliminates the need for complex extraction sequences or specialized scavenger resins to remove heavy metal residues. These factors collectively contribute to a substantially lower cost of goods sold (COGS) for the final intermediate, providing a competitive edge in pricing negotiations.
- Enhanced Supply Chain Reliability: Reliability is the cornerstone of a resilient supply chain. The starting materials for this synthesis, specifically isatin derivatives and aromatic amines used to generate the imidoyl chlorides, are widely available from multiple global suppliers. This multi-sourcing capability prevents bottlenecks that can occur when relying on single-source proprietary reagents. Additionally, the robustness of the reaction conditions—tolerating air and utilizing standard glassware or reactor materials—means that production can be easily transferred between different manufacturing sites without extensive re-validation. This flexibility ensures continuity of supply even in the face of regional disruptions or logistical challenges.
- Scalability and Environmental Compliance: As regulatory pressures mount regarding heavy metal discharge and solvent waste, this green chemistry approach offers a compliant pathway forward. Iron is non-toxic and environmentally benign compared to other transition metals, simplifying wastewater treatment requirements and reducing the environmental footprint of the manufacturing process. The reaction has been demonstrated to be scalable from gram levels in the lab to potentially multi-kilogram batches in pilot plants without loss of efficiency. This scalability ensures that as demand for the final API grows, the supply of the intermediate can be ramped up quickly to meet market needs without requiring capital-intensive new infrastructure.
Frequently Asked Questions (FAQ)
To assist technical teams in evaluating this technology for their specific projects, we have compiled answers to common inquiries regarding the practical application of this patent. These insights address concerns about substrate compatibility, purification challenges, and the specific advantages of the catalytic system employed.
Q: What are the primary advantages of using FeCl3 over traditional catalysts for quinazolinone synthesis?
A: The use of ferric chloride (FeCl3) offers significant economic and operational advantages compared to precious metal catalysts or harsh acidic conditions. It is inexpensive, commercially abundant, and demonstrates high catalytic efficiency for this specific decarbonylative cyclization, reducing overall production costs while maintaining high functional group tolerance.
Q: Can this synthesis method accommodate diverse substituents on the aromatic ring?
A: Yes, the method described in patent CN111675662B exhibits excellent substrate scope. It successfully tolerates various substituents including alkyl groups (methyl), halogens (fluorine, bromine, chlorine), and electron-withdrawing groups (nitro) at ortho-, meta-, or para-positions, allowing for the synthesis of a wide library of analogues for SAR studies.
Q: Is this process suitable for large-scale industrial manufacturing?
A: The protocol is designed with scalability in mind. The use of robust reagents like isatin and trifluoroethylimidoyl chloride, combined with a simple work-up procedure involving filtration and column chromatography, facilitates expansion from gram-level laboratory synthesis to multi-kilogram commercial production without requiring specialized high-pressure equipment.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Quinazolinone Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in accelerating drug development timelines. Our team of expert chemists has thoroughly analyzed the methodology presented in CN111675662B and possesses the technical expertise to implement this iron-catalyzed route effectively. We understand that moving from a patent concept to commercial reality requires more than just following a recipe; it demands rigorous process optimization and quality control. Our facilities are equipped to handle complex heterocyclic synthesis, ensuring that every batch meets stringent purity specifications required by top-tier pharmaceutical companies.
We invite you to leverage our capabilities to secure your supply of these valuable building blocks. Whether you require custom synthesis for early-stage discovery or scaled production for clinical trials, our technical procurement team is ready to assist. Contact us today to request a Customized Cost-Saving Analysis tailored to your project needs. We encourage potential partners to reach out for specific COA data and route feasibility assessments to see how we can support your next breakthrough in oncology or anti-infective therapy.