Advanced Palladium-Catalyzed Synthesis of 2-Trifluoromethyl Quinazolinone Derivatives for Commercial Scale-up
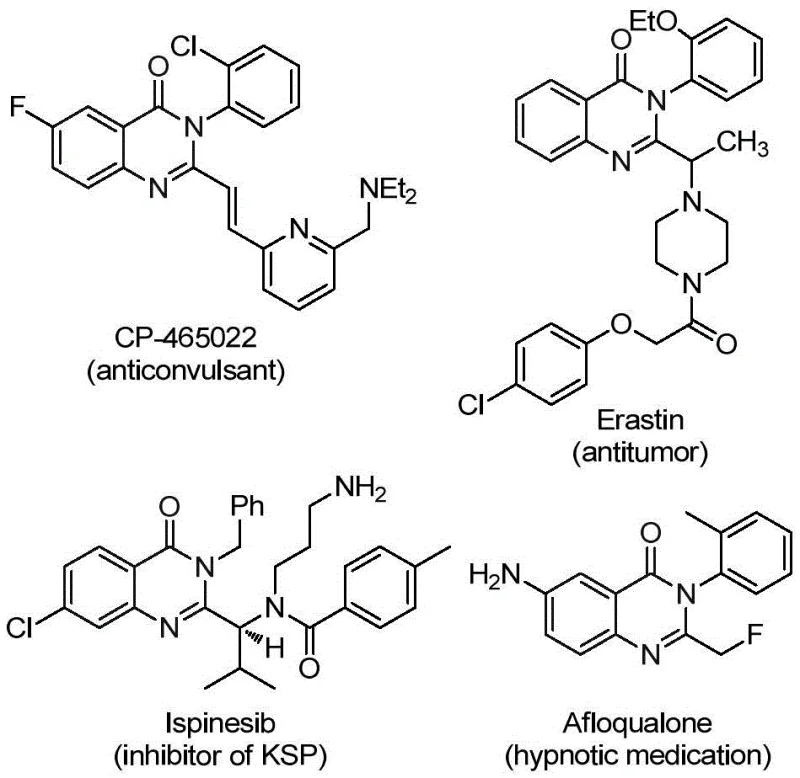
The pharmaceutical industry continuously seeks robust synthetic pathways for heterocyclic scaffolds that possess significant biological activity. Patent CN112125856A introduces a groundbreaking preparation method for 2-trifluoromethyl substituted quinazolinone derivatives, a class of compounds renowned for their presence in numerous therapeutic agents ranging from anticancer drugs to anticonvulsants. The introduction of a trifluoromethyl group into the quinazolinone core significantly enhances physicochemical properties such as metabolic stability, lipophilicity, and bioavailability, making these intermediates highly coveted in modern drug discovery pipelines. This technical insight report analyzes the novel palladium-catalyzed carbonylation tandem reaction detailed in the patent, highlighting its potential to revolutionize the manufacturing of high-purity pharmaceutical intermediates by replacing hazardous gaseous reagents with safer solid surrogates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of 2-trifluoromethyl substituted quinazolinone derivatives has been plagued by significant operational hazards and inefficiencies that hinder large-scale commercial adoption. Traditional methodologies often rely on the direct use of carbon monoxide gas, a colorless, odorless, and highly toxic substance that requires specialized high-pressure equipment and stringent safety protocols to handle safely. Furthermore, existing literature describes routes involving cyclization reactions with ethyl trifluoroacetate or trifluoroacetic anhydride, which frequently suffer from harsh reaction conditions, expensive pre-activated substrates, and limited functional group tolerance. These conventional approaches often result in lower yields and generate substantial waste, creating bottlenecks in the supply chain for reliable agrochemical intermediate supplier and pharma partners who require consistent quality and safety standards.
The Novel Approach
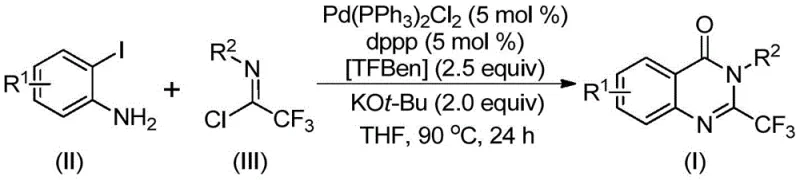
In stark contrast, the methodology disclosed in CN112125856A offers a transformative solution by employing 1,3,5-tricarboxylate phenol ester (TFBen) as a solid carbon monoxide substitute. This innovation effectively eliminates the need for handling toxic carbon monoxide gas, thereby drastically simplifying the reactor setup and reducing the regulatory burden associated with hazardous gas storage. The process utilizes readily available starting materials such as trifluoroethylimidoyl chloride and o-iodoaniline, which are compatible with a wide range of substituents including halogens and alkyl groups. By shifting from gas-phase carbonylation to a solid-surrogate mediated transition metal catalysis, this approach not only enhances operator safety but also improves the overall atom economy and scalability of the synthesis, addressing critical pain points for procurement managers focused on cost reduction in electronic chemical manufacturing and pharmaceutical sectors.
Mechanistic Insights into Pd-Catalyzed Carbonylation Tandem Reaction
The core of this synthetic breakthrough lies in a sophisticated palladium-catalyzed carbonylation tandem reaction mechanism that ensures high efficiency and selectivity. The reaction initiates with a base-promoted intermolecular carbon-nitrogen bond coupling between the o-iodoaniline and trifluoroethylimidoyl chloride, facilitated by potassium tert-butoxide, to form a trifluoroacetamidine derivative intermediate. Subsequently, the palladium catalyst inserts into the carbon-iodine bond of the aromatic ring, generating a reactive divalent palladium species. Under heating conditions at 90°C, the solid TFBen decomposes to release carbon monoxide in situ, which then inserts into the carbon-palladium bond to form a crucial acyl-palladium intermediate. This sequence avoids the accumulation of free CO gas in the headspace, maintaining a controlled reaction environment that is essential for reproducible commercial scale-up of complex polymer additives and fine chemicals.
Following the formation of the acyl-palladium species, the base promotes the formation of a palladium-nitrogen bond, leading to the construction of a seven-membered ring palladium intermediate. The final step involves a reductive elimination process that releases the target 2-trifluoromethyl substituted quinazolinone derivative and regenerates the active palladium catalyst for the next cycle. This mechanistic pathway is particularly advantageous for impurity control, as the tandem nature of the reaction minimizes the formation of side products often associated with stepwise syntheses. The ability to tolerate various substituents on the aryl ring, such as nitro, chloro, or methyl groups, without compromising the catalytic cycle demonstrates the robustness of this method for producing high-purity OLED material and pharmaceutical precursors with diverse structural requirements.
How to Synthesize 2-Trifluoromethyl Quinazolinone Efficiently
Implementing this synthesis route requires precise control over reaction parameters to maximize yield and purity while maintaining safety standards. The protocol dictates the combination of specific molar ratios of palladium catalyst, ligand, and base in an aprotic solvent like tetrahydrofuran. The detailed standardized synthesis steps below outline the exact procedure for combining the solid CO surrogate with the amine and imidoyl chloride substrates, ensuring that technical teams can replicate the high conversion rates reported in the patent data. Adhering to these guidelines is critical for achieving the desired product specifications required by global regulatory bodies.
- Combine palladium catalyst, dppp ligand, potassium tert-butoxide, TFBen solid CO source, trifluoroethylimidoyl chloride, and o-iodoaniline in an organic solvent such as THF.
- Heat the reaction mixture to 90°C and maintain stirring for a duration between 16 to 30 hours to ensure complete conversion.
- Upon completion, filter the mixture, mix with silica gel, and purify the crude product via column chromatography to isolate the target derivative.
Commercial Advantages for Procurement and Supply Chain Teams
From a strategic procurement perspective, the adoption of this solid-carbonyl surrogate technology offers profound advantages that extend beyond mere chemical yield. The elimination of toxic carbon monoxide gas from the supply chain removes the need for specialized gas cylinder logistics, high-pressure reactor certifications, and extensive safety monitoring systems, which translates directly into substantial cost savings in facility operations. For supply chain heads, this means a more resilient production process that is less susceptible to disruptions caused by hazardous material transport regulations or safety incidents. The use of cheap and easily obtainable starting materials further stabilizes the raw material supply, ensuring continuous production capability even during market fluctuations.
- Cost Reduction in Manufacturing: The replacement of gaseous carbon monoxide with a solid surrogate significantly reduces the capital expenditure required for safety infrastructure and gas handling equipment. By avoiding the use of expensive pre-activated substrates and harsh conditions typical of older methods, the overall operational expenditure is lowered through simplified workup procedures and reduced energy consumption for pressure maintenance. This qualitative improvement in process efficiency allows manufacturers to offer more competitive pricing structures without compromising on the quality of the high-purity pharmaceutical intermediates delivered to clients.
- Enhanced Supply Chain Reliability: Utilizing stable solid reagents like TFBen instead of compressed gases mitigates risks associated with transportation and storage, leading to a more predictable and reliable supply chain. The broad substrate compatibility ensures that production lines can be quickly adapted to synthesize different derivatives without extensive retooling, thereby reducing lead time for high-purity pharmaceutical intermediates. This flexibility is crucial for meeting the dynamic demands of R&D departments that require rapid access to diverse compound libraries for drug screening programs.
- Scalability and Environmental Compliance: The reaction operates under atmospheric pressure conditions using standard glassware or stainless steel reactors, making the transition from laboratory scale to multi-ton commercial production seamless and straightforward. The avoidance of toxic gas emissions aligns with increasingly stringent environmental regulations, reducing the burden on waste treatment facilities and minimizing the ecological footprint of the manufacturing process. This commitment to green chemistry principles enhances the corporate sustainability profile, appealing to partners who prioritize environmentally responsible sourcing strategies.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this patented synthesis method. These answers are derived directly from the experimental data and beneficial effects described in the patent documentation, providing clarity on reaction scope, safety profiles, and scalability potential. Understanding these details is essential for technical decision-makers evaluating the feasibility of integrating this route into their existing manufacturing portfolios.
Q: What is the primary safety advantage of this synthesis method?
A: The method utilizes 1,3,5-tricarboxylate phenol ester (TFBen) as a solid carbon monoxide substitute, completely avoiding the use of toxic and hazardous carbon monoxide gas.
Q: What are the optimal reaction conditions described in the patent?
A: The reaction is optimally conducted at 90°C in tetrahydrofuran (THF) solvent for 16 to 30 hours, using a molar ratio of o-iodoaniline to trifluoroethylimidoyl chloride of approximately 1:2.
Q: Does this method support diverse substrate scopes?
A: Yes, the protocol demonstrates high compatibility with various substituents on the aryl ring, including halogens, alkyl groups, and nitro groups, allowing for the design of multiple derivative structures.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Quinazolinone Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of adopting advanced synthetic methodologies to meet the evolving needs of the global pharmaceutical market. Our team of expert chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that innovative laboratory processes like the one described in CN112125856A can be successfully translated into industrial reality. We maintain stringent purity specifications and operate rigorous QC labs to guarantee that every batch of 2-trifluoromethyl quinazolinone derivative meets the highest international standards for identity, potency, and impurity profiles.
We invite you to collaborate with us to leverage this cutting-edge technology for your next project. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and comprehensive route feasibility assessments to demonstrate how our manufacturing capabilities can drive efficiency and reliability in your supply chain.