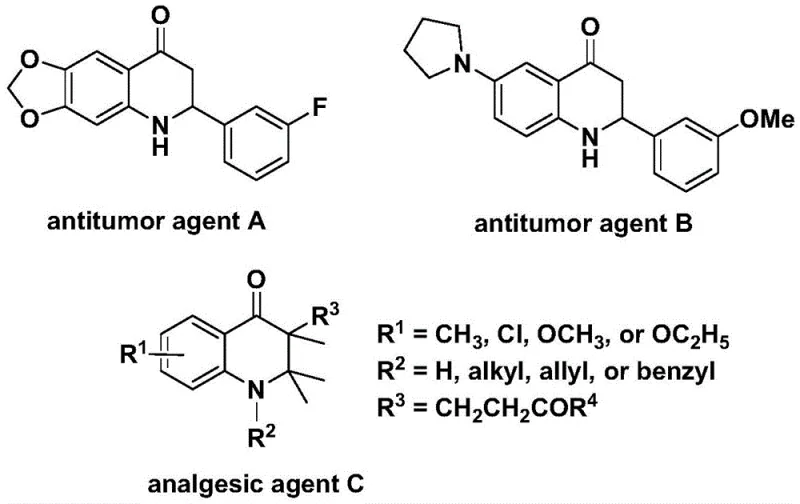
Advanced Palladium-Catalyzed Synthesis of Substituted 2,3-Dihydroquinolone for Commercial Pharmaceutical Production
The patent CN112239456B introduces a groundbreaking palladium-catalyzed carbonylation methodology for synthesizing substituted 2,3-dihydroquinolone compounds, which serve as critical scaffolds in numerous bioactive pharmaceuticals. This innovation addresses longstanding challenges in heterocyclic chemistry by providing a streamlined route to these nitrogen-containing carbonyl structures that are prevalent in anticancer and analgesic agents. The process leverages readily available starting materials—N-pyridine sulfonyl-o-iodoaniline and olefins—under mild reaction conditions that significantly enhance operational safety compared to traditional high-pressure carbonylation techniques. Crucially, the methodology demonstrates exceptional substrate versatility, enabling the synthesis of both 2-aryl and 3-alkyl substituted variants through simple precursor modifications. This flexibility directly supports pharmaceutical R&D teams in rapidly generating diverse compound libraries for structure-activity relationship studies without requiring specialized equipment or hazardous reagents. The patent's emphasis on gram-scale feasibility further underscores its immediate relevance for early-stage drug development programs where reliable intermediate supply is paramount.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional syntheses of 2,3-dihydroquinolone frameworks often rely on multistep sequences involving harsh reaction conditions such as strong acids or high-pressure carbon monoxide environments, which present significant safety hazards and scalability challenges for commercial manufacturing. These approaches typically suffer from poor functional group tolerance, necessitating extensive protection-deprotection strategies that dramatically increase process complexity and reduce overall yield. The requirement for specialized high-pressure reactors creates substantial capital expenditure barriers while limiting production flexibility across different facility types. Furthermore, conventional methods frequently generate complex impurity profiles due to uncontrolled side reactions under aggressive conditions, necessitating costly purification steps that compromise the final product's purity—a critical concern for pharmaceutical intermediates where stringent quality specifications must be met. The limited substrate scope of existing protocols also restricts medicinal chemists' ability to rapidly explore structural analogs during lead optimization phases, ultimately delaying drug discovery timelines and increasing development costs through repeated synthetic route redesigns.
The Novel Approach
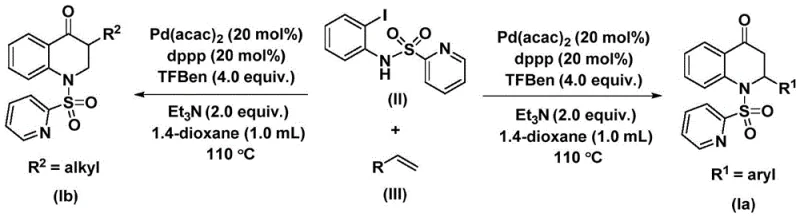
The patented methodology overcomes these limitations through an elegant palladium-catalyzed carbonylation strategy that operates under mild atmospheric pressure conditions at temperatures between 100°C and 120°C. By employing N-pyridine sulfonyl-o-iodoaniline as a directing group precursor and utilizing triethylamine as a base with dioxane as the solvent system, the process achieves exceptional regioselectivity without requiring hazardous reagents or specialized equipment. The catalyst system comprising Pd(acac)₂ (20 mol%) and dppp ligand (20 mol%) demonstrates remarkable efficiency across diverse olefin substrates including aryl derivatives with electron-donating or withdrawing substituents as well as alkyl and silyl variants. This broad substrate compatibility enables pharmaceutical manufacturers to access structurally diverse intermediates from a single platform technology while maintaining consistent product quality. The simplified workup procedure—consisting merely of filtration followed by standard column chromatography—eliminates complex metal removal steps required in alternative methods, thereby reducing processing time and minimizing potential contamination risks that could compromise final API purity specifications.
Mechanistic Insights into Palladium-Catalyzed Carbonylation
The reaction mechanism proceeds through a well-defined catalytic cycle initiated by oxidative addition of palladium into the carbon-nitrogen bond of N-pyridine sulfonyl-o-iodoaniline to form an aryl palladium intermediate. Carbon monoxide released from the triethylamine additive inserts into this intermediate to generate an acyl palladium species, which subsequently coordinates with the olefin substrate to form a palladium alkyl complex through migratory insertion. This key step determines the regioselectivity of the cyclization process and is facilitated by the directing effect of the pyridine sulfonyl group that positions the reaction trajectory favorably. Reductive elimination then occurs to yield the substituted 2,3-dihydroquinolone product while regenerating the active palladium catalyst for subsequent cycles. The precise control over this sequence prevents undesired β-hydride elimination pathways that commonly lead to impurity formation in related catalytic systems, thereby ensuring high product fidelity essential for pharmaceutical applications where even trace impurities can trigger regulatory rejection during quality assessment.
Impurity control is further enhanced by the reaction's inherent chemoselectivity—functional groups such as halogens (F, Cl), alkoxy groups (OMe), and alkyl substituents remain unaffected throughout the process due to the mild reaction conditions and selective catalyst activation. The N-pyridine sulfonyl moiety serves as both a directing group and a protecting element that prevents unwanted side reactions at the nitrogen center while facilitating clean cyclization. Post-reaction purification via standard column chromatography effectively removes any residual catalyst or minor byproducts without requiring specialized techniques like chelation or extraction that might introduce new contaminants. This streamlined approach consistently delivers products meeting pharmaceutical-grade purity requirements as evidenced by HRMS verification showing mass accuracy within ±0.0015 Da across multiple compound variants, thereby eliminating costly reprocessing steps that would otherwise impact commercial viability.
How to Synthesize Substituted 2,3-Dihydroquinolone Efficiently
This innovative synthesis route represents a significant advancement in manufacturing substituted 2,3-dihydroquinolone intermediates by combining operational simplicity with exceptional chemical efficiency. The methodology eliminates the need for specialized high-pressure equipment while maintaining high yields across diverse substrate classes through its carefully optimized catalytic system. By utilizing commercially available starting materials and standard laboratory apparatus, this process dramatically reduces technical barriers to implementation while ensuring consistent product quality suitable for pharmaceutical applications. Detailed standardized synthesis steps are provided below to facilitate seamless technology transfer from laboratory to pilot plant scale.
- Combine N-pyridine sulfonyl-o-iodoaniline (II), olefin (III), Pd(acac)₂ catalyst (20 mol%), dppp ligand (20 mol%), TFBen additive (4.0 equiv.), and triethylamine (2.0 equiv.) in anhydrous dioxane solvent under inert atmosphere
- Heat the reaction mixture to 110°C in a sealed vessel and maintain for 48 hours with continuous stirring to ensure complete carbonylation and cyclization
- Perform post-reaction processing by filtration through silica gel followed by column chromatography purification to isolate the substituted 2,3-dihydroquinolone product with high purity
Commercial Advantages for Procurement and Supply Chain Teams
This patented methodology delivers substantial value across procurement and supply chain functions by addressing critical pain points in pharmaceutical intermediate sourcing. The use of readily available commercial reagents and standard processing equipment significantly reduces supply chain vulnerability compared to traditional approaches requiring specialized materials or custom-built reactors. The simplified process flow minimizes production bottlenecks while enhancing manufacturing flexibility across different facility types—a crucial advantage for global pharmaceutical companies managing complex multi-site production networks. These operational improvements directly translate to more reliable delivery schedules and reduced risk of supply disruption that could impact critical drug development timelines.
- Cost Reduction in Manufacturing: The elimination of expensive transition metal removal procedures and high-pressure reactor requirements substantially lowers capital expenditure while reducing operational costs through simplified processing workflows. The use of commercially available catalysts and ligands at optimized loadings minimizes raw material expenses without compromising reaction efficiency. Streamlined purification protocols eliminate multiple processing steps typically required in conventional syntheses, resulting in significant labor and resource savings throughout the manufacturing cycle while maintaining consistent product quality.
- Enhanced Supply Chain Reliability: The reliance on widely accessible starting materials with established global supply networks ensures consistent availability regardless of regional market fluctuations or geopolitical disruptions. The process's compatibility with standard manufacturing equipment enables rapid technology transfer between production sites without requiring specialized infrastructure investments. This flexibility allows for dynamic allocation of production capacity across different facilities based on real-time demand patterns while maintaining consistent quality standards through the well-defined reaction parameters specified in the patent documentation.
- Scalability and Environmental Compliance: The methodology's demonstrated scalability from gram-scale laboratory validation to potential multi-ton production aligns with current industry requirements for seamless technology transfer. The absence of hazardous reagents or extreme reaction conditions significantly reduces environmental impact while simplifying waste stream management compared to traditional approaches. The simplified process flow minimizes energy consumption through lower temperature operation and eliminates complex solvent recovery systems required in alternative methods, thereby supporting corporate sustainability initiatives without compromising manufacturing efficiency.
Frequently Asked Questions (FAQ)
The following questions address key technical and commercial considerations based on detailed analysis of patent CN112239456B's experimental data and implementation requirements. These insights reflect practical experience in scaling similar catalytic processes for pharmaceutical intermediate production while addressing common concerns raised by procurement and R&D stakeholders during technology evaluation.
Q: How does this palladium-catalyzed method improve impurity control compared to conventional approaches?
A: The Pd(acac)₂/dppp catalyst system enables precise regioselective carbonylation at controlled temperatures (100-120°C), minimizing side reactions that generate impurities. The N-pyridine sulfonyl directing group ensures clean cyclization without requiring harsh conditions that typically produce degradation byproducts in traditional syntheses.
Q: What substrate flexibility does this process offer for pharmaceutical intermediate production?
A: The method accommodates diverse olefin substrates (R = aryl with methyl, tert-butyl, methoxy, fluorine or chlorine substituents; alkyl including linear/branched chains; or silyl groups), allowing customization of the 2,3-dihydroquinolone core for specific API requirements while maintaining high functional group tolerance.
Q: How does the elimination of transition metal removal steps impact commercial scale-up feasibility?
A: The catalytic system's high efficiency (up to 88% yield) and simplified post-treatment (filtration followed by column chromatography) eliminate complex metal scavenging procedures. This reduces processing time and avoids costly purification steps required in conventional methods using less selective catalysts.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Substituted 2,3-Dihydroquinolone Supplier
Our patented methodology represents a significant advancement in synthesizing complex heterocyclic intermediates with exceptional purity profiles required for modern pharmaceutical applications. NINGBO INNO PHARMCHEM brings extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production while maintaining stringent purity specifications through our state-of-the-art QC labs equipped with advanced analytical capabilities. Our dedicated technical teams specialize in adapting this palladium-catalyzed platform to meet specific client requirements while ensuring seamless integration with existing manufacturing processes through comprehensive route feasibility assessments.
Leverage our expertise to optimize your supply chain—request a Customized Cost-Saving Analysis today to evaluate how this technology can enhance your production economics. Contact our technical procurement team to obtain specific COA data and detailed route feasibility assessments tailored to your manufacturing needs.