Scalable Production of 2-Trifluoromethyl Quinazolinones via Iron-Catalyzed Cyclization
Scalable Production of 2-Trifluoromethyl Quinazolinones via Iron-Catalyzed Cyclization
The pharmaceutical and fine chemical industries are constantly seeking more efficient, cost-effective, and environmentally benign routes to synthesize complex heterocyclic scaffolds that serve as the backbone for modern therapeutics. A significant breakthrough in this domain is detailed in patent CN111675662B, which discloses a novel preparation method for 2-trifluoromethyl substituted quinazolinone compounds. Quinazolinones are privileged structures in medicinal chemistry, renowned for their diverse biological activities ranging from anticancer and anticonvulsant to anti-inflammatory and antifungal properties. The strategic introduction of a trifluoromethyl group into these heterocycles further enhances their metabolic stability, lipophilicity, and bioavailability, making them highly desirable candidates for drug development. This patent presents a robust synthetic strategy that overcomes many of the historical limitations associated with trifluoromethylated heterocycle synthesis, offering a pathway that is not only chemically elegant but also commercially viable for large-scale manufacturing.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of quinazolinones bearing a trifluoromethyl functional group has been fraught with significant challenges that hinder their widespread adoption in industrial settings. Traditional methodologies often rely on the cyclization of synthons containing the trifluoromethyl group, such as trifluoroacetic anhydride or ethyl trifluoroacetate, with substrates like anthranilamide, anthranilic acid, or isatoic anhydride. While these methods can yield the desired products, they are frequently plagued by severe reaction conditions that require stringent temperature control or hazardous reagents. Furthermore, the starting materials utilized in these conventional routes are often expensive and not readily available in bulk quantities, driving up the overall cost of goods. Another critical drawback is the narrow substrate scope; many traditional protocols fail to tolerate sensitive functional groups, limiting the chemical diversity accessible to medicinal chemists. Low yields and difficult purification processes due to side reactions further exacerbate the inefficiency, making these routes less attractive for the commercial scale-up of complex pharmaceutical intermediates.
The Novel Approach
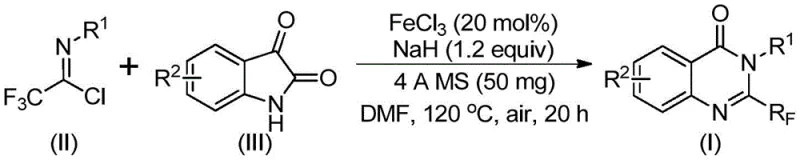
In stark contrast to these legacy methods, the technology described in patent CN111675662B introduces a streamlined and highly efficient approach utilizing readily available starting materials. The core innovation lies in the use of trifluoroethylimidoyl chloride and isatin derivatives as the primary building blocks. This reaction is catalyzed by ferric chloride (FeCl3), an inexpensive and earth-abundant transition metal, in the presence of sodium hydride and 4A molecular sieves. This new methodology eliminates the need for costly precious metal catalysts and harsh conditions, operating effectively in common organic solvents like DMF. The process demonstrates exceptional functional group tolerance, allowing for the incorporation of various substituents such as halogens, alkyls, and nitro groups without compromising yield. By shifting to this iron-catalyzed decarbonylation and cyclization strategy, manufacturers can achieve high conversion rates and simplified post-treatment procedures, significantly enhancing the overall economic feasibility of producing these valuable heterocyclic compounds.
Mechanistic Insights into FeCl3-Catalyzed Cyclization
The success of this synthetic route is underpinned by a sophisticated yet practical mechanistic pathway driven by the iron catalyst. The reaction initiates with the formation of carbon-nitrogen bonds between the trifluoroethylimidoyl chloride and the isatin substrate, facilitated by the base sodium hydride. This initial step generates a trifluoroacetamidine intermediate, which serves as the precursor for the subsequent cyclization. The ferric chloride catalyst then plays a pivotal role in promoting a decarbonylation event, effectively removing a carbonyl group and triggering an isomerization that closes the ring to form the quinazolinone core. This iron-mediated process is remarkably efficient, likely proceeding through a coordination complex that lowers the activation energy for the cyclization step. The use of 4A molecular sieves is also critical, as they act as a desiccant to sequester water generated during the reaction or present in the reagents, thereby preventing hydrolysis of the sensitive imidoyl chloride and driving the equilibrium towards the desired product. This mechanistic understanding allows for precise optimization of reaction parameters, ensuring consistent quality and high purity in the final output.
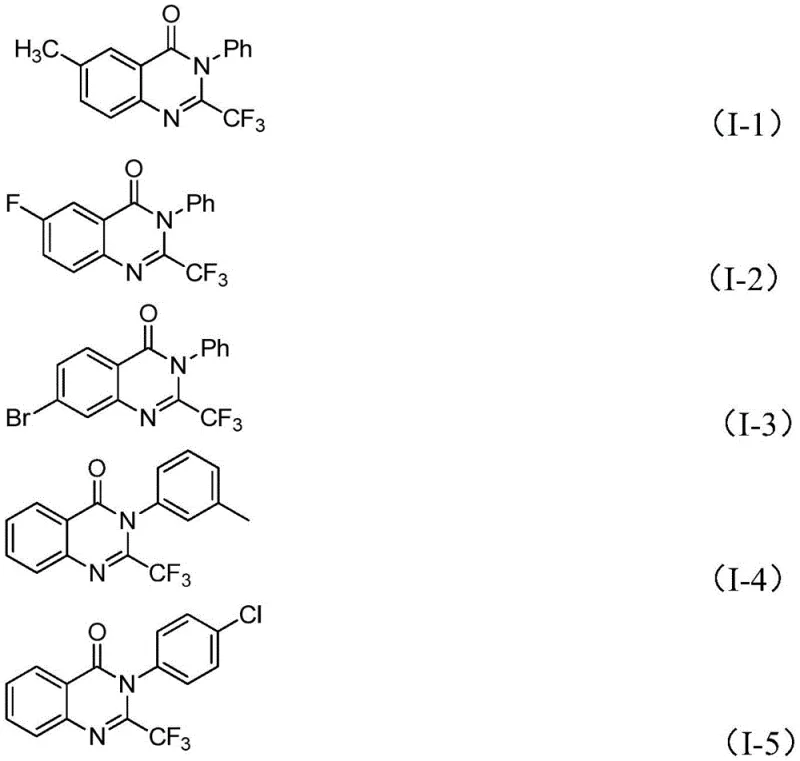
Furthermore, the robustness of this catalytic system is evidenced by its broad substrate compatibility, which is essential for generating diverse libraries of analogs for structure-activity relationship (SAR) studies. The method accommodates various substitutions on both the aryl ring of the imidoyl chloride and the benzene ring of the isatin. Whether the substituents are electron-donating groups like methyl or methoxy, or electron-withdrawing groups like fluorine, bromine, chlorine, or nitro, the reaction proceeds with high efficiency. This versatility is crucial for R&D teams aiming to explore the chemical space around the quinazolinone scaffold to identify potent drug candidates. The ability to synthesize compounds like those shown in the specific examples (I-1 to I-5) with yields reaching as high as 93% underscores the reliability of this mechanism. Such high yields and purity profiles minimize the burden on downstream purification processes, reducing waste and solvent consumption, which aligns perfectly with green chemistry principles and cost-reduction goals in pharmaceutical manufacturing.
How to Synthesize 2-Trifluoromethyl Quinazolinones Efficiently
Implementing this synthesis in a laboratory or pilot plant setting requires adherence to specific operational parameters to maximize yield and safety. The process involves a two-stage heating protocol where the reaction mixture is first maintained at a moderate temperature to facilitate the initial coupling, followed by a higher temperature phase to drive the cyclization to completion. The use of anhydrous conditions and proper stoichiometry of the base and catalyst is essential to prevent side reactions. For detailed operational specifics regarding reagent ratios, solvent volumes, and exact temperature ramps, please refer to the standardized synthesis steps outlined below, which are derived directly from the optimized embodiments of the patent.
- Mix ferric chloride, sodium hydride, 4A molecular sieves, trifluoroethylimidoyl chloride, and isatin in an organic solvent like DMF.
- React the mixture at 40°C for 8-10 hours, then heat to 120°C for an additional 18-20 hours under air.
- Filter the reaction mixture, mix with silica gel, and purify via column chromatography to obtain the final product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the transition to this iron-catalyzed synthesis method represents a strategic opportunity to optimize the supply of critical pharmaceutical intermediates. The shift away from precious metal catalysts and expensive, specialized reagents towards commodity chemicals like ferric chloride and isatin drastically simplifies the sourcing landscape. This change mitigates the risk of supply chain disruptions often associated with rare earth or precious metal dependencies. Moreover, the operational simplicity of the process—requiring standard reactor equipment and common solvents—means that existing manufacturing infrastructure can be utilized without significant capital expenditure on new hardware. The high yields and clean reaction profiles reported in the patent suggest a substantial reduction in raw material waste, directly contributing to lower production costs and a smaller environmental footprint. These factors combined create a resilient and cost-efficient supply chain for high-purity quinazolinone derivatives.
- Cost Reduction in Manufacturing: The replacement of expensive catalysts and complex starting materials with inexpensive, commercially available alternatives like FeCl3 and isatin leads to a direct decrease in the bill of materials. The elimination of costly purification steps required to remove trace precious metals further reduces processing expenses. Additionally, the high atom economy and yield of the reaction minimize the amount of raw material needed per kilogram of finished product, resulting in significant overall cost savings for the manufacturing of API intermediates.
- Enhanced Supply Chain Reliability: By utilizing starting materials that are widely produced and stocked by global chemical suppliers, the risk of raw material shortages is significantly minimized. The robustness of the reaction conditions ensures consistent batch-to-batch quality, reducing the likelihood of production delays caused by failed runs or out-of-specification results. This reliability allows for more accurate forecasting and inventory management, ensuring a steady flow of intermediates to downstream drug formulation units.
- Scalability and Environmental Compliance: The process is inherently scalable, having been demonstrated to work efficiently from gram scales in the lab to potential ton-scale production. The use of iron, a non-toxic and environmentally benign metal, simplifies waste treatment and disposal compared to processes using heavy metals. This alignment with green chemistry principles facilitates easier regulatory approval and compliance with increasingly stringent environmental regulations, smoothing the path for commercial scale-up of complex pharmaceutical intermediates.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production of 2-trifluoromethyl substituted quinazolinones. These answers are grounded in the experimental data and technical specifications provided in the source patent, offering clarity on the feasibility and benefits of this synthetic route for potential partners and stakeholders.
Q: What are the advantages of using FeCl3 over traditional catalysts for quinazolinone synthesis?
A: Using ferric chloride (FeCl3) offers significant cost advantages as it is an abundant, inexpensive base metal compared to precious metal catalysts. Additionally, it demonstrates excellent functional group tolerance and operates under relatively mild conditions, simplifying the purification process and reducing heavy metal contamination risks in the final API intermediate.
Q: Can this synthesis method accommodate diverse substituents on the aromatic ring?
A: Yes, the method exhibits broad substrate scope. It successfully tolerates various substituents including alkyl groups (methyl), halogens (fluorine, bromine, chlorine, iodine), and electron-withdrawing groups (nitro, methoxy) at ortho-, meta-, or para-positions, allowing for the generation of a diverse library of analogs for SAR studies.
Q: Is this process suitable for large-scale industrial manufacturing?
A: Absolutely. The protocol utilizes commercially available starting materials like isatin and trifluoroethylimidoyl chloride, avoids cryogenic conditions, and uses standard solvents like DMF. The robustness of the iron catalysis and the simplicity of the work-up procedure make it highly amenable to scale-up from gram-level laboratory synthesis to multi-ton commercial production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Quinazolinone Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in the drug development pipeline. Our team of expert chemists has thoroughly analyzed the innovative route described in patent CN111675662B and is fully prepared to leverage this technology for your projects. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with precision and consistency. Our state-of-the-art facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications, guaranteeing that every batch of 2-trifluoromethyl quinazolinone we deliver meets the highest industry standards for pharmaceutical applications.
We invite you to collaborate with us to unlock the full potential of this efficient synthesis method. Whether you require custom synthesis for early-stage research or large-scale manufacturing for clinical trials, our technical procurement team is ready to assist. Please contact us today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are eager to provide you with specific COA data and comprehensive route feasibility assessments to demonstrate how our capabilities can accelerate your project timelines and reduce your overall development costs.