Scalable Palladium-Catalyzed Carbonylation for High-Purity Quinazolinone Pharmaceutical Intermediates
Scalable Palladium-Catalyzed Carbonylation for High-Purity Quinazolinone Pharmaceutical Intermediates
The quinazolinone scaffold represents a cornerstone in modern medicinal chemistry, underpinning a vast array of bioactive molecules with potent therapeutic profiles. As illustrated in 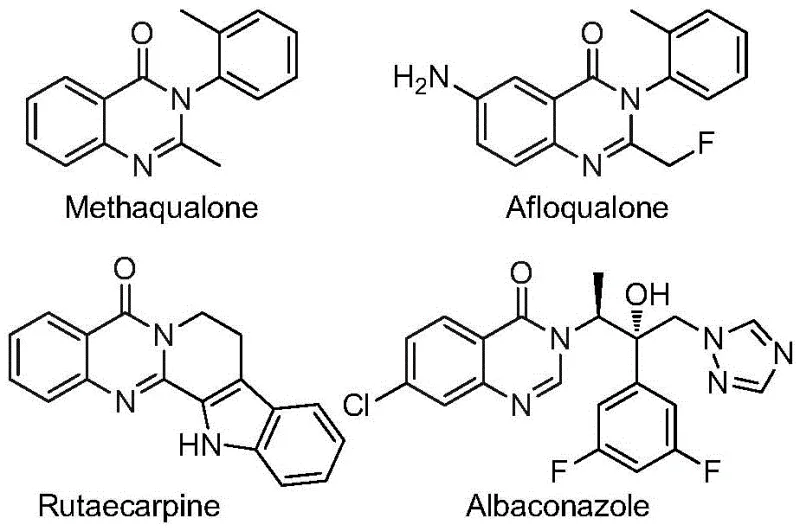 , this heterocyclic core is prevalent in drugs exhibiting antifungal, antibacterial, antiviral, and anticancer activities. The introduction of a trifluoromethyl group further enhances these properties by improving metabolic stability and lipophilicity. Addressing the critical need for efficient access to these valuable scaffolds, the recent technological breakthrough detailed in patent CN112480015B introduces a robust multi-component one-pot method. This innovation specifically targets the synthesis of 2-trifluoromethyl substituted quinazolinones, leveraging a palladium-catalyzed carbonylation cascade that transforms inexpensive nitro compounds into high-value pharmaceutical intermediates with exceptional efficiency.
, this heterocyclic core is prevalent in drugs exhibiting antifungal, antibacterial, antiviral, and anticancer activities. The introduction of a trifluoromethyl group further enhances these properties by improving metabolic stability and lipophilicity. Addressing the critical need for efficient access to these valuable scaffolds, the recent technological breakthrough detailed in patent CN112480015B introduces a robust multi-component one-pot method. This innovation specifically targets the synthesis of 2-trifluoromethyl substituted quinazolinones, leveraging a palladium-catalyzed carbonylation cascade that transforms inexpensive nitro compounds into high-value pharmaceutical intermediates with exceptional efficiency.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of the quinazolinone ring system has been plagued by significant synthetic hurdles that hinder cost-effective manufacturing. Traditional pathways often rely on the use of high-pressure carbon monoxide gas, which necessitates specialized, expensive reactor equipment and poses severe safety risks in a production environment. Furthermore, existing protocols frequently demand pre-activated substrates, such as 2-bromoformylaniline or acid anhydrides, which are not only costly but also generate substantial chemical waste during their own preparation. Other methods involving iron or ruthenium catalysts often suffer from narrow substrate scope, harsh reaction conditions, and mediocre yields, making them unsuitable for the rigorous purity standards required in reliable pharmaceutical intermediate supplier networks. These limitations collectively drive up the cost of goods sold (COGS) and complicate the supply chain for complex heterocyclic APIs.
The Novel Approach
The methodology disclosed in CN112480015B fundamentally reshapes this landscape by employing a transition metal palladium-catalyzed carbonylation cascade using nitro compounds as the nitrogen source. As depicted in the general reaction scheme in 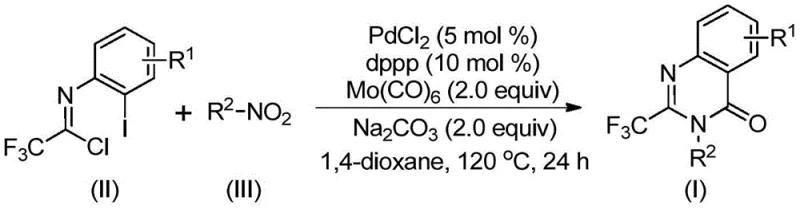 , this approach utilizes trifluoroethylimidoyl chloride and nitro compounds in the presence of Mo(CO)6 as a safe, solid carbon monoxide surrogate. This eliminates the need for high-pressure gas infrastructure entirely. The reaction proceeds in a single pot at 120°C in common solvents like 1,4-dioxane, demonstrating remarkable functional group tolerance. By bypassing the need for pre-reduced amines or activated halides, this novel route achieves cost reduction in API manufacturing through simplified logistics and reduced step counts, while maintaining high reaction efficiency and yield.
, this approach utilizes trifluoroethylimidoyl chloride and nitro compounds in the presence of Mo(CO)6 as a safe, solid carbon monoxide surrogate. This eliminates the need for high-pressure gas infrastructure entirely. The reaction proceeds in a single pot at 120°C in common solvents like 1,4-dioxane, demonstrating remarkable functional group tolerance. By bypassing the need for pre-reduced amines or activated halides, this novel route achieves cost reduction in API manufacturing through simplified logistics and reduced step counts, while maintaining high reaction efficiency and yield.
Mechanistic Insights into Pd-Catalyzed Carbonylation Cascade
The success of this synthesis lies in the intricate orchestration of a multi-step catalytic cycle initiated by the reduction of the nitro group. Mechanistically, the reaction likely commences with the reduction of the nitro compound to the corresponding amine by Mo(CO)6, which serves a dual role as both the CO source and the reductant. This generated amine then undergoes a base-promoted intermolecular coupling with trifluoroethylimidoyl chloride to form a trifluoroacetamidine derivative in situ. Subsequently, the palladium catalyst, stabilized by the dppp ligand, inserts into the carbon-iodine bond of the imidoyl chloride moiety, forming a key divalent palladium intermediate. The thermal decomposition of Mo(CO)6 releases carbon monoxide, which inserts into the carbon-palladium bond to generate an acyl-palladium species.
Following CO insertion, the intramolecular nucleophilic attack by the nitrogen atom promotes the formation of a seven-membered cyclic palladium intermediate. This cyclization step is critical for establishing the quinazolinone core. Finally, reductive elimination releases the desired 2-trifluoromethyl substituted quinazolinone product and regenerates the active palladium catalyst. This mechanism ensures high chemoselectivity, minimizing the formation of side products such as ureas or simple amides that often plague amidine syntheses. The broad substrate compatibility, evidenced by the successful synthesis of diverse derivatives shown in 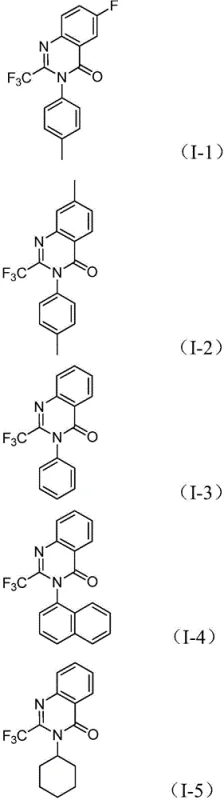 , confirms that electron-withdrawing and electron-donating groups on the aromatic rings are well-tolerated, allowing for the precise tuning of electronic properties required for drug discovery campaigns.
, confirms that electron-withdrawing and electron-donating groups on the aromatic rings are well-tolerated, allowing for the precise tuning of electronic properties required for drug discovery campaigns.
How to Synthesize 2-Trifluoromethyl Quinazolinone Efficiently
Implementing this synthesis requires careful attention to the stoichiometry of the carbonyl source and the choice of ligand to maximize turnover. The protocol dictates the use of palladium chloride and 1,3-bis(diphenylphosphino)propane (dppp) in a specific molar ratio to ensure stable catalytic activity throughout the 16 to 30-hour reaction window. Sodium carbonate acts as the base to neutralize the HCl byproduct generated during the amidine formation, driving the equilibrium forward. The detailed standardized synthesis steps, including precise reagent quantities and workup procedures validated by the patent examples, are outlined below for technical replication.
- Combine palladium chloride, dppp ligand, sodium carbonate, Mo(CO)6, trifluoroethylimidoyl chloride, and nitro compound in an organic solvent like dioxane.
- Heat the reaction mixture to 120°C and stir for 16 to 30 hours to allow the carbonylation cascade and cyclization to proceed.
- Upon completion, filter the mixture, mix with silica gel, and purify via column chromatography to isolate the target quinazolinone.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement strategists and supply chain directors, the shift from high-pressure gas methods to this solid-state carbonylation protocol offers transformative economic and logistical benefits. The primary driver of value is the substitution of expensive, hazardous, or pre-activated starting materials with commodity chemicals. Nitro compounds are ubiquitous in the fine chemical market, ensuring a stable and competitive supply base that is less susceptible to the volatility seen with specialized organometallic reagents. Furthermore, the elimination of high-pressure CO cylinders removes a major regulatory and safety burden from the manufacturing site, significantly lowering overhead costs related to safety compliance and equipment maintenance.
- Cost Reduction in Manufacturing: The economic advantage of this process is derived principally from the atom economy and the price differential of the raw materials. By utilizing nitro compounds instead of pre-synthesized amines or acid anhydrides, the number of synthetic steps prior to the main reaction is effectively reduced to zero. This consolidation of steps translates directly into lower labor costs, reduced solvent consumption, and decreased waste disposal fees. Additionally, the use of Mo(CO)6 as a stoichiometric CO source avoids the capital expenditure associated with purchasing and certifying high-pressure autoclaves, allowing production to occur in standard glass-lined reactors.
- Enhanced Supply Chain Reliability: Supply chain resilience is bolstered by the commercial availability of the key reagents. Trifluoroethylimidoyl chloride and various substituted nitrobenzenes are produced by multiple global suppliers, mitigating the risk of single-source dependency. The robustness of the reaction conditions—specifically the tolerance to moisture and air compared to sensitive organolithium or Grignard reagents often used in alternative routes—further ensures consistent batch-to-batch quality. This reliability is crucial for maintaining continuous production schedules for high-purity pharmaceutical intermediates without unexpected delays caused by reagent instability or sourcing bottlenecks.
- Scalability and Environmental Compliance: From an environmental and scaling perspective, the one-pot nature of this reaction minimizes the generation of intermediate waste streams. The ability to run the reaction in 1,4-dioxane, a solvent with established recovery and recycling protocols in the industry, supports green chemistry initiatives. The patent data indicates that the reaction can be scaled to gram levels with maintained efficiency, suggesting a smooth path to kilogram and ton-scale production. The simplified post-treatment, involving filtration and standard column chromatography or crystallization, reduces the complexity of downstream processing, facilitating faster throughput and commercial scale-up of complex heterocyclic intermediates.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and optimization of this synthesis route. These answers are derived directly from the experimental data and mechanistic understanding provided in the patent documentation, ensuring that R&D teams have accurate information for process development.
Q: What are the advantages of using nitro compounds over traditional amines in this synthesis?
A: Nitro compounds are significantly cheaper and more readily available than pre-activated amines or acid anhydrides. This method utilizes them directly, reducing raw material costs and simplifying the supply chain for large-scale production.
Q: How does this method improve safety compared to traditional carbonylation?
A: Traditional methods often require high-pressure carbon monoxide gas, which poses significant safety risks. This patent utilizes Mo(CO)6 as a solid CO surrogate, eliminating the need for high-pressure gas equipment and enhancing operational safety.
Q: Is this process suitable for industrial scale-up?
A: Yes, the patent explicitly states the method can be expanded to the gram level and beyond. The one-pot nature reduces unit operations, and the use of common solvents like dioxane facilitates standard industrial processing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Quinazolinone Supplier
At NINGBO INNO PHARMCHEM, we recognize the strategic importance of efficient heterocycle synthesis in accelerating drug development timelines. Our technical team has thoroughly analyzed the capabilities of the Pd-catalyzed carbonylation method described in CN112480015B and is prepared to leverage this technology for your specific project needs. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory discovery to industrial manufacturing is seamless. Our facilities are equipped with stringent purity specifications and rigorous QC labs capable of verifying the structural integrity and impurity profiles of complex fluorinated intermediates.
We invite you to collaborate with us to optimize this route for your specific target molecules. By partnering with our technical procurement team, you can request a Customized Cost-Saving Analysis that compares this novel one-pot method against your current supply chain. Contact us today to obtain specific COA data for related quinazolinone derivatives and comprehensive route feasibility assessments tailored to your commercial goals.