Advanced FeCl3-Catalyzed Synthesis of 2-Trifluoromethyl Quinazolinones for Commercial Scale-Up
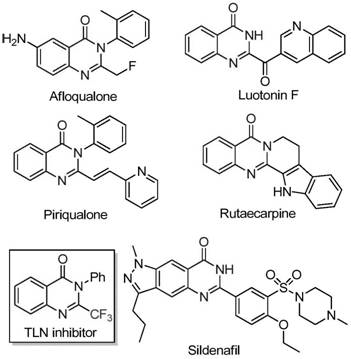
The pharmaceutical and fine chemical industries are constantly seeking robust synthetic methodologies to access bioactive heterocyclic scaffolds efficiently. A recent breakthrough detailed in patent CN111675662B introduces a highly effective preparation method for 2-trifluoromethyl substituted quinazolinone compounds, which are critical motifs in modern drug design. These nitrogen-containing fused ring systems are renowned for their diverse biological activities, including anti-cancer, anticonvulsant, and antifungal properties, making them indispensable building blocks for advanced therapeutic agents. The strategic introduction of the trifluoromethyl group significantly enhances the electronegativity, metabolic stability, and lipophilicity of the target molecules, thereby improving their bioavailability and pharmacokinetic profiles. This technological advancement addresses the longstanding challenges associated with synthesizing these complex structures, offering a pathway that balances high yield with operational simplicity. For R&D directors and procurement specialists, understanding this novel route is essential for securing a reliable pharmaceutical intermediates supplier capable of delivering high-quality materials for next-generation drug development pipelines.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of quinazolinone derivatives bearing trifluoromethyl functional groups has been plagued by significant technical and economic hurdles that hinder large-scale adoption. Traditional methodologies often rely on the cyclization of synthons containing the trifluoromethyl group with substrates such as anthranilamide or isatoic anhydride, which frequently necessitates the use of expensive and hazardous reagents like trifluoroacetic anhydride. These conventional routes are typically characterized by severe reaction conditions, requiring extreme temperatures or pressures that pose safety risks and increase energy consumption in a manufacturing setting. Furthermore, the substrate scope in older methods is often narrow, limiting the ability to introduce diverse functional groups without compromising yield or purity. The reliance on costly starting materials and the generation of difficult-to-remove impurities result in elevated production costs and extended lead times, creating bottlenecks for supply chain heads who require consistent and economical access to high-purity OLED material or pharmaceutical precursors. Consequently, there is an urgent industry demand for a more sustainable and cost-effective alternative.
The Novel Approach
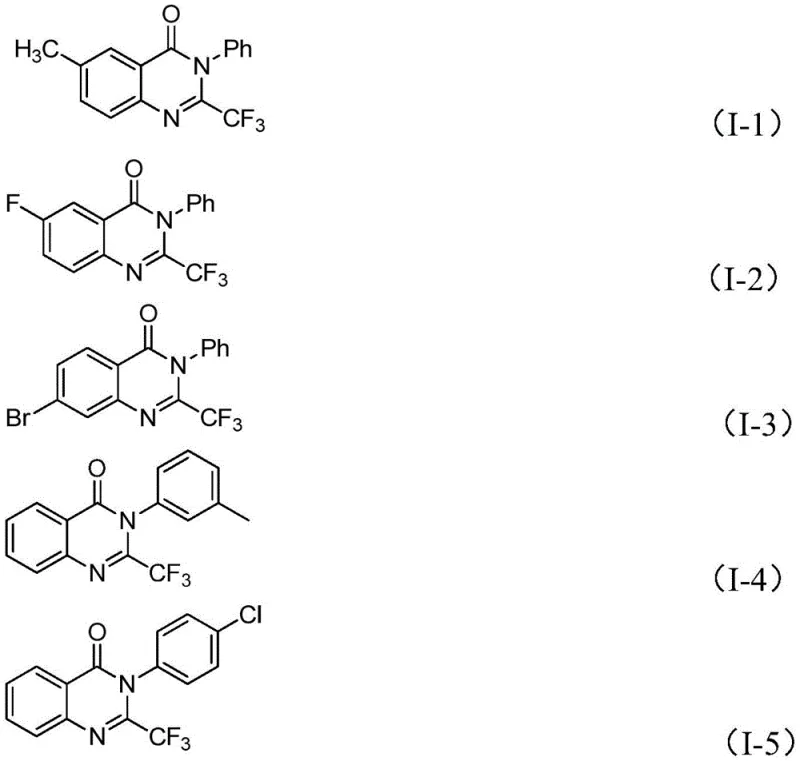
In stark contrast to legacy techniques, the novel approach outlined in the patent utilizes readily available trifluoroethylimidoyl chloride and isatin as starting raw materials, catalyzed by inexpensive iron salts. This method represents a paradigm shift in cost reduction in fine chemical manufacturing by replacing precious metal catalysts with abundant ferric chloride, drastically lowering the raw material expenditure. The reaction proceeds through a series of cyclization steps under relatively mild conditions, initially at 40°C and subsequently heated to 120°C, which minimizes thermal degradation of sensitive functional groups. The operational simplicity is further enhanced by the use of common organic solvents like DMF and the inclusion of 4A molecular sieves to manage moisture, ensuring reproducible results across different batches. By enabling the synthesis of various substituted quinazolinones with high efficiency and broad applicability, this route provides a scalable solution that meets the rigorous demands of commercial scale-up of complex polymer additives and pharmaceutical intermediates alike.
Mechanistic Insights into FeCl3-Catalyzed Cyclization
The core of this synthetic innovation lies in the intricate mechanistic pathway facilitated by the iron catalyst, which drives the formation of the quinazolinone core with remarkable precision. The reaction initiates with an alkali-promoted formation of carbon-nitrogen bonds between the trifluoroethylimidoyl chloride and the isatin substrate, generating a trifluoroacetamidine intermediate. This step is crucial as it sets the stage for the subsequent iron-catalyzed decarbonylation and cyclization reactions that ultimately isomerize the intermediate into the desired 2-trifluoromethyl-substituted quinazolinone structure. The presence of ferric chloride acts as a Lewis acid, activating the carbonyl groups and facilitating the nucleophilic attacks required for ring closure without the need for exotic ligands or inert atmospheres. Understanding this mechanism is vital for R&D teams aiming to optimize reaction parameters for specific derivatives, as it highlights the tolerance of the system to various electronic environments on the aromatic rings. The ability to control the regioselectivity and ensure complete conversion through this catalytic cycle underscores the robustness of the chemistry, providing a solid foundation for process validation and regulatory filing.
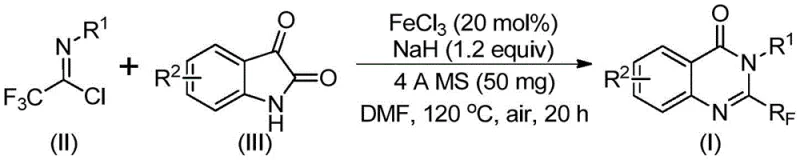
Furthermore, the impurity control mechanism inherent in this process is designed to minimize the formation of side products that often complicate downstream purification. The specific molar ratios of ferric chloride to sodium hydride, optimized at approximately 0.2 to 1.2, ensure that the base is sufficient to drive the deprotonation steps without causing excessive decomposition of the sensitive imidoyl chloride species. The use of 4A molecular sieves plays a pivotal role in scavenging trace water that could otherwise hydrolyze the reactive intermediates, thereby maintaining high reaction integrity throughout the 18 to 20 hour heating period. Post-reaction processing involves standard filtration and column chromatography, techniques that are well-established in industrial settings for achieving stringent purity specifications. This level of control over the chemical environment ensures that the final product meets the rigorous quality standards required for high-purity pharmaceutical intermediates, reducing the risk of batch rejection and ensuring supply continuity for downstream customers.
How to Synthesize 2-Trifluoromethyl Quinazolinone Efficiently
To implement this synthesis effectively, operators must adhere to precise procedural guidelines that maximize yield while maintaining safety standards in the laboratory or plant. The process begins with the careful addition of ferric chloride and sodium hydride to a reaction vessel containing the organic solvent, followed by the introduction of the molecular sieves and substrates under controlled stirring. It is imperative to monitor the temperature profile closely, maintaining the initial phase at 40°C for 8 to 10 hours before ramping up to 120°C to drive the cyclization to completion. Detailed standardized synthesis steps see the guide below.
- Prepare the reaction mixture by adding ferric chloride, sodium hydride, 4A molecular sieves, trifluoroethylimidoyl chloride, and isatin into an organic solvent such as DMF.
- Initiate the reaction at 40°C for 8-10 hours, then increase the temperature to 120°C and maintain for 18-20 hours under air atmosphere.
- Upon completion, filter the mixture, mix with silica gel, and perform column chromatography purification to isolate the final 2-trifluoromethyl substituted quinazolinone compound.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, this patented methodology offers transformative benefits for procurement managers and supply chain leaders focused on optimizing operational expenditures and mitigating risk. The substitution of expensive noble metal catalysts with ferric chloride results in substantial cost savings, as iron is one of the most abundant and affordable transition metals available in the global market. This shift not only reduces the direct cost of goods sold but also simplifies the waste management process, as the disposal of iron residues is less regulated and costly compared to heavy metals like palladium or platinum. Additionally, the use of commercially available starting materials such as isatin and aromatic amines ensures a stable supply chain, reducing the vulnerability to shortages of specialized reagents that can disrupt production schedules. The robustness of the reaction conditions allows for flexible manufacturing planning, enabling producers to respond quickly to fluctuating market demands without compromising on quality or delivery timelines.
- Cost Reduction in Manufacturing: The elimination of precious metal catalysts and the use of inexpensive reagents like sodium hydride and DMF significantly lower the overall production cost per kilogram. By avoiding complex ligand systems and inert gas protections, the process reduces utility consumption and equipment requirements, leading to drastic simplification of the manufacturing workflow. These efficiencies translate into competitive pricing for the final intermediates, allowing pharmaceutical companies to allocate more resources towards clinical development and market expansion rather than raw material procurement.
- Enhanced Supply Chain Reliability: The reliance on bulk chemicals that are widely produced globally ensures a consistent and reliable supply of raw materials, minimizing the risk of production stoppages due to sourcing issues. The simplified post-treatment process, which avoids specialized extraction techniques, shortens the manufacturing cycle time, allowing for faster turnaround from order to delivery. This agility is crucial for maintaining inventory levels and meeting the just-in-time delivery expectations of major multinational corporations in the healthcare sector.
- Scalability and Environmental Compliance: The reaction demonstrates excellent scalability, having been validated from gram-scale experiments to potential multi-ton production runs without loss of efficiency. The reduced generation of hazardous waste and the absence of toxic heavy metals align with increasingly strict environmental regulations, facilitating easier permitting and compliance auditing. This green chemistry approach enhances the corporate sustainability profile of manufacturers, making them preferred partners for eco-conscious buyers seeking to reduce their carbon footprint.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation and benefits of this synthesis technology. These answers are derived directly from the experimental data and beneficial effects described in the patent documentation, providing clarity for stakeholders evaluating this technology for adoption. Understanding these details is key to making informed decisions about integrating this route into existing production portfolios.
Q: What are the primary advantages of using FeCl3 over traditional catalysts for quinazolinone synthesis?
A: The use of ferric chloride (FeCl3) offers substantial cost advantages compared to precious metal catalysts often used in heterocyclic synthesis. It eliminates the need for expensive heavy metal removal steps, simplifying downstream processing and reducing overall production costs while maintaining high reaction efficiency.
Q: How does this method improve substrate tolerance for pharmaceutical applications?
A: This protocol demonstrates excellent functional group tolerance, accommodating various substituents such as halogens, alkyl, and methoxy groups on the aromatic rings. This versatility allows for the efficient generation of diverse libraries of quinazolinone derivatives essential for drug discovery and development without requiring extensive protection-deprotection strategies.
Q: Is this synthesis method suitable for large-scale industrial production?
A: Yes, the method utilizes readily available starting materials like isatin and trifluoroethylimidoyl chloride, and operates under relatively mild conditions. The simplicity of the post-treatment process, involving standard filtration and chromatography, supports scalability from gram-level laboratory synthesis to multi-ton commercial manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Quinazolinone Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of accessing advanced synthetic technologies to drive innovation in the pharmaceutical and fine chemical sectors. As a premier CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project transitions smoothly from the laboratory bench to full-scale manufacturing. Our state-of-the-art facilities are equipped with rigorous QC labs and adhere to stringent purity specifications, guaranteeing that every batch of 2-trifluoromethyl quinazolinone delivered meets the highest international standards. We are committed to leveraging our technical expertise to optimize this iron-catalyzed route, providing you with a secure and efficient source of high-value intermediates for your drug development programs.
We invite you to collaborate with us to explore the full potential of this cost-effective synthesis method for your specific application needs. Please contact our technical procurement team to request a Customized Cost-Saving Analysis tailored to your volume requirements. We are ready to provide specific COA data and comprehensive route feasibility assessments to demonstrate how our capabilities can enhance your supply chain resilience and accelerate your time to market.