Scalable Synthesis of Molnupiravir Intermediates: A Technical Breakthrough for Commercial Manufacturing
Scalable Synthesis of Molnupiravir Intermediates: A Technical Breakthrough for Commercial Manufacturing
The global demand for effective antiviral therapeutics has placed immense pressure on the supply chains of critical active pharmaceutical ingredients (APIs), particularly for broad-spectrum agents like Molnupiravir (EIDD-2801). Patent CN113956312B, published in late 2022, introduces a transformative preparation method that addresses the longstanding bottlenecks in synthesizing this vital nucleoside analog. This technical disclosure outlines a robust, two-step synthetic pathway that replaces hazardous reagents and complex purification steps with safer, solid-state alternatives and efficient biphasic reactions. For R&D directors and procurement strategists, this patent represents a pivotal shift towards a more sustainable and economically viable manufacturing paradigm. By fundamentally altering the oximation and deprotection stages, the methodology ensures product purity exceeding 99.8%, a critical benchmark for regulatory compliance in raw material drug production. The following analysis dissects the chemical innovations and their profound implications for the reliable API intermediate supplier landscape.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior to this innovation, the industrial synthesis of Molnupiravir was plagued by significant operational hazards and inefficiencies that hindered cost reduction in pharmaceutical manufacturing. As illustrated in the legacy pathways, earlier methods often relied on multi-step sequences involving oily intermediates that necessitated tedious column chromatography for purification. This reliance on chromatographic separation is a major bottleneck for scale-up, as it consumes vast quantities of silica gel and solvents, drastically increasing waste generation and processing time. Furthermore, alternative routes utilized aqueous hydroxylamine solutions for the critical oximation step. Aqueous hydroxylamine is classified as an explosive hazardous compound, posing severe safety risks during transportation, storage, and feeding in large reactors. These safety concerns, coupled with the inability to effectively remove hydrolytic impurities in subsequent steps, resulted in products with poor purity profiles and single impurity levels exceeding 0.1%, rendering them unsuitable for high-grade pharmaceutical applications without extensive and costly rework.
The Novel Approach
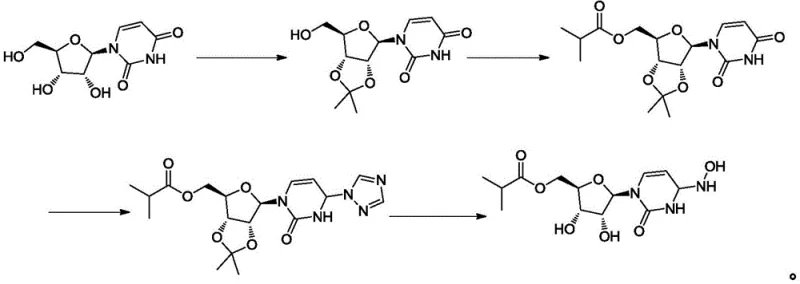
The methodology disclosed in CN113956312B offers a decisive break from these conventional constraints by introducing a streamlined, safety-first protocol. The core innovation lies in the substitution of dangerous liquid reagents with stable solid salts and the implementation of a controlled biphasic deprotection system. Instead of using explosive aqueous hydroxylamine, the new process employs hydroxylamine sulfate or hydroxylamine hydrochloride in conjunction with sodium acetate within an alcohol-water solvent system. This switch not only mitigates explosion risks but also enhances reaction stability and reproducibility. Following the formation of the key intermediate (Formula II), the deprotection is conducted in a dichloromethane or chloroform medium with hydrochloric acid at strictly controlled low temperatures. This specific solvent choice and temperature control create a biphasic environment that kinetically favors the desired deprotection while suppressing unwanted hydrolysis. The result is a process that is inherently safer, environmentally friendlier due to reduced solvent usage, and capable of delivering high-purity antiviral intermediates directly through crystallization rather than chromatography.
Mechanistic Insights into Solid-State Oximation and Biphasic Deprotection
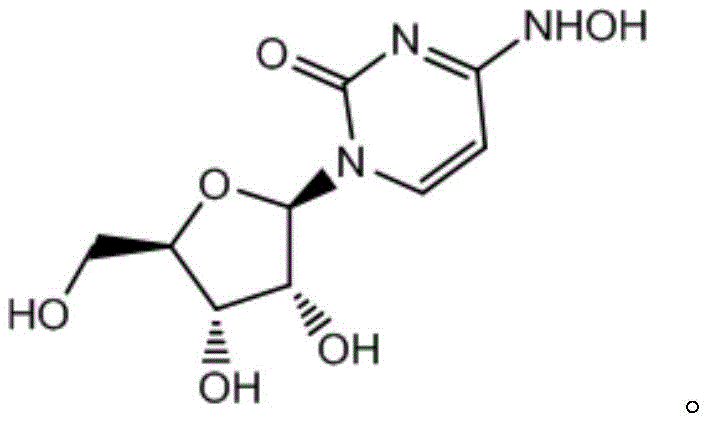
The chemical elegance of this new route is rooted in its precise control over reaction kinetics and phase behavior. In the first step, the conversion of the protected uridine derivative (Formula I) to the oxime intermediate (Formula II) is driven by the in situ generation of reactive hydroxylamine species from solid salts. The presence of sodium acetate acts as a buffer, maintaining an optimal pH that facilitates nucleophilic attack on the carbonyl group while preventing the degradation of the sensitive ribose moiety. Operating at temperatures between 70-80°C ensures complete conversion within a reasonable timeframe (17-20 hours) without inducing thermal decomposition. The use of solid reagents ensures a consistent stoichiometric feed, eliminating the variability often seen with aged or unstable liquid reagents. This consistency is crucial for maintaining batch-to-batch uniformity, a key requirement for commercial scale-up of complex nucleoside analogs.
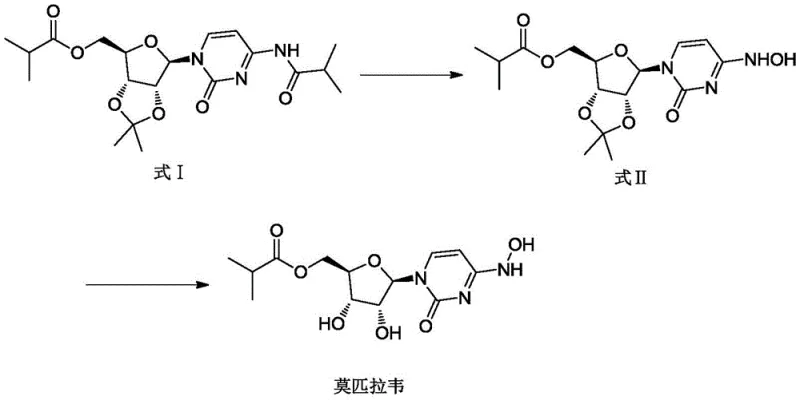
In the second critical stage, the deprotection of Formula II to yield Molnupiravir, the mechanism relies heavily on solubility differences and phase partitioning to manage impurities. The reaction generates two primary potential impurities: unreacted intermediate (Impurity 1) and the hydrolyzed byproduct (Impurity 2). By conducting the reaction in a halogenated solvent like dichloromethane with aqueous hydrochloric acid, a two-phase system is established. The desired Molnupiravir product exhibits high solubility in the aqueous phase once formed, while Impurity 1 remains preferentially dissolved in the organic phase. This allows for a simple liquid-liquid separation to remove the starting material residue. Furthermore, the hydrolytic Impurity 2, which is highly water-soluble, is effectively washed away during the subsequent recrystallization step using purified water. This mechanistic understanding of phase distribution allows manufacturers to achieve purity levels greater than 99.8% without the need for preparative HPLC or column chromatography, significantly simplifying the downstream processing train.
How to Synthesize Molnupiravir Efficiently
The implementation of this synthesis route requires strict adherence to the specified reaction conditions to maximize yield and minimize impurity formation. The process begins with the dissolution of the protected precursor in an alcohol solvent such as ethanol or isopropanol, followed by the addition of water and the solid reagent mixture. Precise temperature control during the heating and cooling phases is essential to ensure proper crystallization of the intermediate. The subsequent deprotection step demands even tighter thermal regulation, specifically maintaining the reaction mixture between 0-5°C during acid addition to prevent exothermic runaway and hydrolytic degradation. The workup involves a strategic pH adjustment using weak bases like ammonia or sodium bicarbonate to neutralize the acid before extraction.
- React Formula (I) with sodium acetate and solid hydroxylamine sulfate or hydrochloride in an alcohol-water solvent system at 70-80°C to form Formula (II).
- Treat Formula (II) with hydrochloric acid in dichloromethane or chloroform at 0-5°C to effect deprotection while minimizing hydrolysis.
- Purify the crude product via aqueous recrystallization, leveraging the water solubility of hydrolytic impurities to achieve >99.8% purity.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this patented methodology translates into tangible operational improvements and risk mitigation. The elimination of column chromatography is perhaps the most significant economic driver, as it removes a major cost center associated with silica gel consumption, solvent recovery, and extended processing times. This simplification directly contributes to cost reduction in pharmaceutical manufacturing by shortening the overall cycle time and reducing the facility footprint required for production. Moreover, the shift from hazardous liquid hydroxylamine to stable solid salts dramatically lowers the regulatory burden and insurance costs associated with handling explosive materials. This enhances the overall safety profile of the manufacturing site, ensuring business continuity and reducing the risk of shutdowns due to safety incidents. The robustness of the purification strategy, which relies on crystallization rather than complex separations, ensures that the process is readily transferable from pilot plants to multi-ton commercial reactors without loss of efficiency.
- Cost Reduction in Manufacturing: The removal of column chromatography steps eliminates the need for expensive silica gel and large volumes of elution solvents, leading to substantial savings in raw material costs and waste disposal fees. The use of common, recyclable solvents like ethanol and dichloromethane further optimizes the cost structure, making the process economically superior to legacy routes that rely on specialized purification techniques.
- Enhanced Supply Chain Reliability: By utilizing stable, non-hazardous solid reagents instead of explosive aqueous solutions, the supply chain becomes more resilient and less prone to logistical disruptions caused by hazardous material transport regulations. The simplified process flow reduces the dependency on specialized equipment and skilled operators for chromatography, allowing for more flexible production scheduling and faster response to market demand fluctuations.
- Scalability and Environmental Compliance: The process is designed with green chemistry principles in mind, significantly reducing the E-factor (mass of waste per mass of product) by avoiding chromatographic waste streams. The ability to use water for recrystallization minimizes the release of organic solvents into the environment, facilitating easier compliance with increasingly stringent environmental regulations and supporting corporate sustainability goals.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis route. Understanding these details is crucial for technical teams evaluating the feasibility of adopting this method for their own production lines. The answers are derived directly from the experimental data and mechanistic insights provided in the patent documentation, ensuring accuracy and relevance for decision-makers.
Q: Why is solid hydroxylamine sulfate preferred over aqueous hydroxylamine in this synthesis?
A: Solid hydroxylamine salts eliminate the safety risks associated with transporting and handling explosive aqueous hydroxylamine solutions, significantly improving industrial safety profiles.
Q: How does the new process control hydrolytic impurities compared to prior art?
A: By utilizing a biphasic reaction system with hydrochloric acid in dichloromethane at low temperatures (0-5°C), the process kinetically suppresses the formation of hydrolytic impurities, which are subsequently removed via water recrystallization.
Q: Is this synthesis route suitable for large-scale commercial production?
A: Yes, the method avoids column chromatography and uses standard solvents and solid reagents, making it highly scalable, cost-effective, and environmentally friendly for metric-ton manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Molnupiravir Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of securing a stable supply of high-quality antiviral intermediates in the current global health landscape. Our technical team has thoroughly analyzed the pathway described in CN113956312B and possesses the expertise to implement this advanced synthesis strategy effectively. We boast extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that our clients receive consistent quality regardless of order volume. Our state-of-the-art facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications, guaranteeing that every batch of Molnupiravir intermediate meets the >99.8% purity threshold required for API manufacturing.
We invite pharmaceutical partners to collaborate with us to leverage this cost-effective and safe production technology. By engaging with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage potential partners to contact us directly to obtain specific COA data and comprehensive route feasibility assessments, ensuring that your supply chain is built on a foundation of technical excellence and commercial reliability.