Scalable Iron-Catalyzed Synthesis of 2-Trifluoromethyl Quinazolinones for Pharmaceutical Applications
Scalable Iron-Catalyzed Synthesis of 2-Trifluoromethyl Quinazolinones for Pharmaceutical Applications
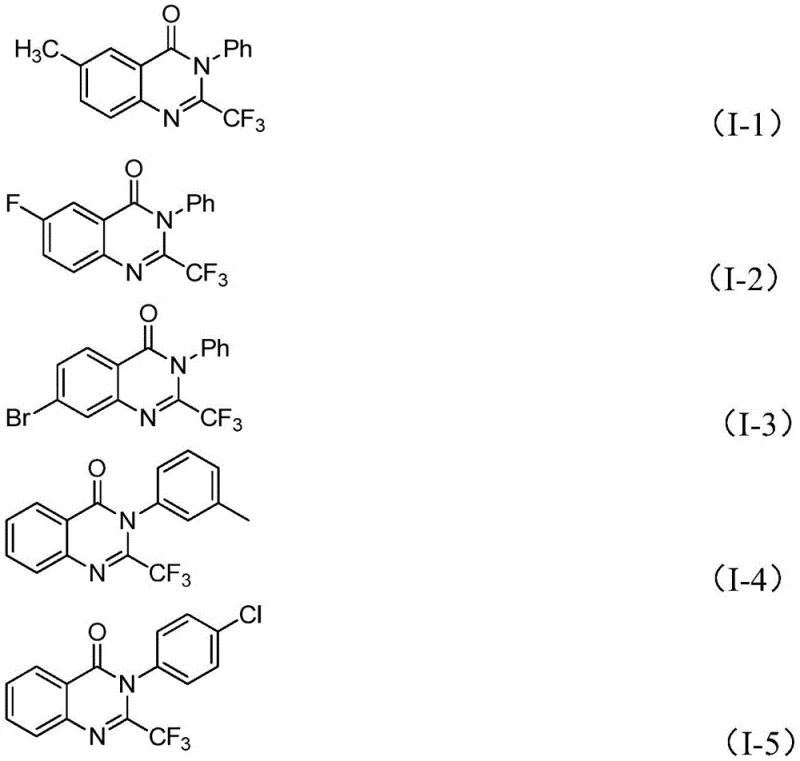
The development of efficient synthetic routes for nitrogen-containing heterocycles remains a cornerstone of modern medicinal chemistry, particularly for scaffolds exhibiting potent biological activity. A recent breakthrough detailed in patent CN111675662A introduces a robust methodology for preparing 2-trifluoromethyl-substituted quinazolinone compounds, a class of molecules renowned for their anticancer, anticonvulsant, and anti-inflammatory properties. This innovation addresses critical bottlenecks in the supply chain for high-purity pharmaceutical intermediates by leveraging an inexpensive iron-catalyzed tandem cyclization strategy. By utilizing readily available starting materials such as isatin and trifluoroethylimidoyl chloride, this process eliminates the reliance on costly precious metal catalysts and harsh reaction conditions that have historically plagued the synthesis of these valuable heterocyclic frameworks.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of the quinazolinone core bearing a trifluoromethyl group has been fraught with significant chemical and economic challenges. Traditional literature methods often rely on the cyclization of anthranilamides or anthranilic acids with expensive trifluoromethyl synthons such as trifluoroacetic anhydride or ethyl trifluoroacetate. These conventional pathways are frequently hampered by severe reaction conditions that require extreme temperatures or pressures, leading to poor atom economy and difficult downstream processing. Furthermore, the substrate scope in these older methodologies is often narrow, failing to tolerate sensitive functional groups which limits the structural diversity accessible to medicinal chemists. The combination of low yields, expensive reagents, and limited versatility makes these legacy processes unsuitable for the cost-sensitive and high-volume demands of the global pharmaceutical supply chain.
The Novel Approach
In stark contrast, the novel approach disclosed in the patent utilizes a tandem cyclization reaction catalyzed by ferric chloride, offering a streamlined and economically superior alternative. This method employs isatin and trifluoroethylimidoyl chloride as primary building blocks, which are not only commercially abundant but also significantly cheaper than traditional synthons. The reaction proceeds through a sophisticated cascade involving carbon-nitrogen bond formation followed by decarbonylation, effectively installing the trifluoromethyl group with high precision. As illustrated in the specific examples below, this methodology supports a wide array of substituents, allowing for the rapid generation of diverse libraries of quinazolinone derivatives essential for drug discovery campaigns.
Mechanistic Insights into FeCl3-Catalyzed Cyclization
The mechanistic elegance of this transformation lies in the dual role of the reaction components in facilitating a complex rearrangement under relatively mild thermal conditions. The process initiates with a base-promoted intermolecular coupling between the trifluoroethylimidoyl chloride and the isatin nitrogen, forming a transient trifluoroacetamidine intermediate. Subsequently, the ferric chloride catalyst activates the system for a crucial decarbonylation step, which drives the cyclization forward to form the fused heterocyclic ring. This iron-mediated pathway avoids the formation of stable byproducts that typically plague non-catalytic thermal cyclizations, thereby ensuring a cleaner reaction profile and simplifying the purification workload for process chemists.
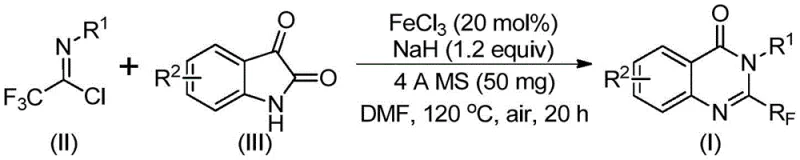
From an impurity control perspective, the use of 4A molecular sieves in the reaction mixture plays a pivotal role in maintaining anhydrous conditions, which is critical for the stability of the acid chloride starting material and the efficiency of the sodium hydride base. The specific stoichiometry of ferric chloride to sodium hydride (optimized at roughly 0.2:1.2 molar ratio) ensures that the basicity is sufficient to drive the initial coupling without causing excessive decomposition of the sensitive trifluoromethyl moiety. This precise balance allows the reaction to proceed with high conversion rates, minimizing the presence of unreacted starting materials and reducing the burden on downstream chromatographic separation steps, ultimately resulting in a higher quality final active pharmaceutical ingredient intermediate.
How to Synthesize 2-Trifluoromethyl Quinazolinones Efficiently
The operational simplicity of this synthesis makes it highly attractive for both laboratory scale-up and eventual commercial manufacturing. The protocol involves a straightforward one-pot procedure where the catalyst, base, and drying agents are combined with the substrates in a polar aprotic solvent such as DMF. The reaction profile utilizes a two-stage temperature gradient, starting at a moderate 40°C to facilitate the initial coupling, followed by heating to 120°C to drive the energetically demanding decarbonylation and cyclization to completion. For detailed standardized operating procedures and safety guidelines regarding the handling of sodium hydride and high-temperature reactions, please refer to the technical documentation provided below.
- Mix ferric chloride catalyst, sodium hydride base, and 4A molecular sieves in anhydrous DMF solvent under inert atmosphere.
- Add trifluoroethylimidoyl chloride and isatin substrates to the reaction mixture and stir at 40°C for 8-10 hours.
- Heat the reaction mixture to 120°C for 18-20 hours to complete the cyclization, then purify via column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the transition to this iron-catalyzed methodology represents a strategic opportunity to optimize the cost structure of quinazolinone-based drug programs. The substitution of expensive precious metal catalysts with commodity-grade ferric chloride drastically reduces the raw material cost per kilogram of the final intermediate. Additionally, the elimination of complex multi-step sequences in favor of a tandem one-pot reaction significantly shortens the overall manufacturing cycle time, allowing for faster response to market demands and reduced inventory holding costs. The robustness of the reaction conditions also implies a lower risk of batch failure, enhancing the reliability of supply for critical clinical trial materials.
- Cost Reduction in Manufacturing: The economic benefits of this process are driven primarily by the replacement of costly reagents with inexpensive, bulk-available chemicals. Ferric chloride is a fraction of the cost of palladium or rhodium catalysts often used in similar C-N bond formations, and the starting materials like isatin are produced on a massive industrial scale globally. Furthermore, the high atom economy of the tandem reaction minimizes waste generation, which translates to lower disposal costs and a reduced environmental footprint, aligning with modern green chemistry mandates without compromising on yield or purity.
- Enhanced Supply Chain Reliability: Sourcing risks are significantly mitigated because the key reagents—isatin and substituted aromatic amines (precursors to the imidoyl chloride)—are established commodities with multiple qualified suppliers worldwide. Unlike specialized fluorinating agents that may have long lead times or single-source dependencies, the inputs for this synthesis are resilient to market fluctuations. This diversity in the supply base ensures continuity of supply even during global logistical disruptions, providing a secure foundation for long-term commercial production agreements and preventing costly delays in drug development timelines.
- Scalability and Environmental Compliance: The process has been demonstrated to be scalable from milligram to gram levels with consistent performance, indicating a clear path to multi-kilogram and ton-scale production. The use of DMF as a solvent, while requiring careful recovery, is well-understood in industrial settings with established recycling protocols. Moreover, the absence of heavy metal residues in the final product simplifies the regulatory filing process, as there is no need for extensive and expensive metal scavenging steps to meet strict ICH guidelines for elemental impurities in pharmaceutical substances, thereby accelerating time-to-market.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These answers are derived directly from the experimental data and scope defined in the patent literature, providing a factual basis for evaluating the feasibility of this route for your specific project needs. Understanding these nuances is essential for R&D teams planning pilot runs and for procurement teams assessing vendor capabilities.
Q: What are the advantages of using FeCl3 over traditional catalysts for quinazolinone synthesis?
A: Using ferric chloride (FeCl3) offers significant cost advantages over precious metal catalysts like palladium or rhodium. It is inexpensive, readily available, and demonstrates high efficiency in promoting the tandem cyclization and decarbonylation steps required for this transformation.
Q: Can this synthesis method tolerate various functional groups on the substrate?
A: Yes, the method exhibits excellent functional group tolerance. Substrates with electron-donating groups (like methyl, methoxy) and electron-withdrawing groups (like halogens, nitro) on both the aryl ring of the imidoyl chloride and the isatin core are compatible, yielding products with high purity.
Q: Is this process suitable for large-scale industrial production?
A: The process is designed for scalability. It utilizes cheap starting materials like isatin and avoids complex purification steps beyond standard column chromatography. The reaction conditions are robust, making it viable for scaling from gram-level laboratory synthesis to multi-kilogram commercial production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Quinazolinone Supplier
As the demand for fluorinated heterocycles continues to surge in the development of next-generation therapeutics, partnering with a manufacturer who possesses deep technical expertise in C-H activation and cyclization chemistry is vital. NINGBO INNO PHARMCHEM stands at the forefront of this field, leveraging advanced catalytic technologies to deliver complex intermediates with exceptional purity profiles. Our facility is equipped with extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can support your needs from early-stage discovery through to full-scale commercial launch. Our rigorous QC labs employ state-of-the-art analytical instrumentation to guarantee that every batch meets stringent purity specifications, minimizing the risk of downstream processing issues.
We invite you to engage with our technical team to explore how this innovative iron-catalyzed route can be integrated into your supply chain to drive efficiency and reduce costs. By requesting a Customized Cost-Saving Analysis, you can gain a clear understanding of the economic impact of switching to this methodology for your specific target molecule. We encourage you to contact our technical procurement team today to obtain specific COA data for our catalog compounds and to discuss route feasibility assessments tailored to your unique project requirements.