Advanced Catalytic Synthesis of Trifluoromethyl Dihydrofuran Amines for Commercial Scale-Up
Advanced Catalytic Synthesis of Trifluoromethyl Dihydrofuran Amines for Commercial Scale-Up
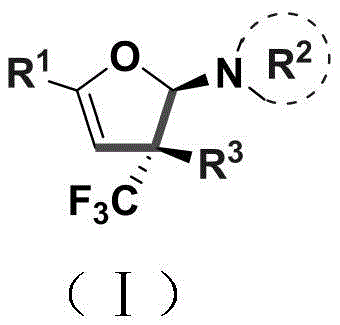
The landscape of heterocyclic chemistry is constantly evolving to meet the rigorous demands of modern drug discovery, particularly regarding the incorporation of fluorine motifs which enhance metabolic stability and bioavailability. Patent CN110922369B introduces a significant breakthrough in this domain by disclosing a novel preparation method for trifluoromethyl-substituted dihydrofuran amine compounds. These structures are not merely academic curiosities; they represent critical scaffolds found in numerous bioactive natural products and pharmaceutical agents. The core innovation lies in the ability to construct these complex five-membered rings bearing a quaternary carbon stereocenter through a streamlined, catalytic process. This technology addresses the longstanding challenge of introducing trifluoromethyl groups into heterocycles from simple acyclic precursors, a transformation that has historically been plagued by difficult reaction conditions and poor selectivity. By leveraging a copper-catalyzed cyclization strategy, this method offers a robust pathway for generating high-value intermediates that are essential for the development of next-generation agrochemicals and APIs.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of 2,3-dihydrofuran rings, especially those substituted with electron-withdrawing trifluoromethyl groups, has relied on methodologies that are often impractical for large-scale manufacturing. Traditional approaches frequently necessitate the use of stoichiometric amounts of expensive transition metals, which not only drives up raw material costs but also creates significant downstream purification burdens due to heavy metal residue limits in pharmaceutical products. Furthermore, many existing protocols operate under harsh thermal conditions or require highly sensitive reagents that exhibit limited functional group tolerance. This lack of chemoselectivity often leads to complex impurity profiles, forcing R&D teams to invest excessive time in optimizing protection-deprotection sequences. The difficulty in constructing quaternary carbon centers with high stereoselectivity from acyclic substrates further compounds these issues, making the reliable supply of such intermediates a bottleneck in many drug development pipelines.
The Novel Approach
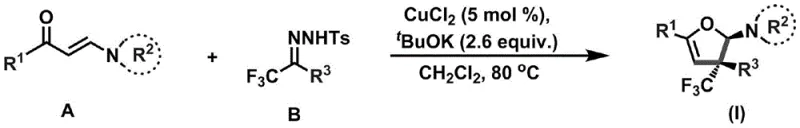
In stark contrast to these legacy methods, the technology described in CN110922369B utilizes a highly efficient catalytic cycle driven by copper chloride. The process involves the reaction of an enaminone (Compound A) with a trifluoromethylhydrazone (Compound B) in the presence of a base and a catalytic amount of copper species. This approach dramatically lowers the barrier to entry for synthesizing these complex motifs by operating at moderate temperatures ranging from 80°C to 100°C. The use of a catalytic system rather than stoichiometric reagents fundamentally changes the economic equation, reducing the consumption of precious metals and simplifying the workup procedure. Moreover, the reaction demonstrates exceptional substrate scope, accommodating a wide variety of aryl, alkyl, and heterocyclic substituents without compromising yield or selectivity. This versatility makes it an ideal candidate for library synthesis and rapid analog generation in medicinal chemistry programs.
Mechanistic Insights into CuCl2-Catalyzed Cyclization
The mechanistic elegance of this transformation lies in its ability to orchestrate a cascade of bond-forming events that result in the simultaneous construction of the furan ring and the installation of the trifluoromethyl group. The reaction initiates with the activation of the trifluoromethylhydrazone by the base, likely generating a diazo or carbene-like intermediate that is subsequently intercepted by the copper catalyst. This metal-carbene species then engages with the electron-rich enaminone double bond, facilitating a cycloaddition or insertion event that closes the five-membered ring. Crucially, the steric and electronic environment provided by the ligands and the substrate geometry ensures the formation of the quaternary carbon stereocenter with high fidelity. This level of stereocontrol is paramount for pharmaceutical applications where the biological activity is often strictly dependent on the three-dimensional arrangement of atoms. The tolerance for diverse functional groups suggests that the catalytic cycle is robust against potential side reactions such as beta-hydride elimination or premature hydrolysis.
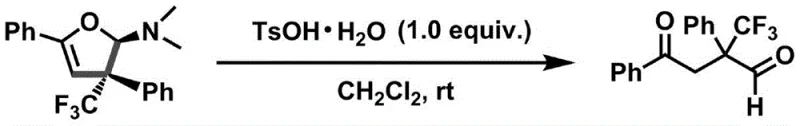
Beyond the primary cyclization, the utility of these dihydrofuran amines is further amplified by their capacity for downstream derivatization. As detailed in the patent, the resulting amine functionality can be hydrolyzed under mild acidic conditions to yield trifluoromethyl-containing 1,4-dicarbonyl compounds. This transformation is quantitative and proceeds at room temperature, indicating that the dihydrofuran ring acts as a masked 1,4-dicarbonyl equivalent. Such 1,4-dicarbonyl systems are versatile building blocks for the synthesis of pyrroles, furans, and other heterocycles prevalent in drug molecules. The ability to access these scaffolds from a common intermediate streamlines the synthetic route, allowing chemists to diverge late in the synthesis to access multiple distinct chemical spaces. This modularity is a key advantage for process chemists looking to maximize the value of a single synthetic platform across multiple projects.
How to Synthesize Trifluoromethyl Dihydrofuran Amine Efficiently
The operational simplicity of this synthesis makes it highly attractive for both laboratory scale-up and industrial production. The protocol requires standard equipment and readily available reagents, minimizing the need for specialized infrastructure. The reaction is typically conducted in dichloromethane, a common solvent that facilitates easy handling and workup. Following the reaction period, the removal of insoluble salts by filtration and subsequent concentration yields a crude product that can be purified via standard silica gel chromatography. For those looking to implement this methodology, the following guide outlines the critical parameters for success, ensuring high purity and yield while maintaining safety standards.
- Mix enaminone (Compound A), trifluoromethylhydrazone (Compound B), CuCl2 catalyst, and potassium tert-butoxide base in dichloromethane under inert gas.
- Heat the reaction mixture to 80-100°C and stir for 48-72 hours to facilitate the cyclization.
- Filter insoluble solids, concentrate the solution under reduced pressure, and purify via column chromatography to isolate the target dihydrofuran amine.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this catalytic methodology presents a compelling value proposition centered on cost efficiency and supply reliability. By shifting from stoichiometric metal reagents to a catalytic system, the direct material costs associated with metal consumption are drastically reduced. Furthermore, the mild reaction conditions mitigate the risks associated with thermal runaways, enhancing process safety and reducing the engineering controls required for manufacturing. The broad substrate scope implies that a single manufacturing line could potentially be utilized to produce a wide array of different analogs simply by swapping the starting enaminone or hydrazone, thereby maximizing asset utilization and flexibility. This adaptability is crucial in a market where demand for specific intermediates can fluctuate rapidly based on clinical trial outcomes.
- Cost Reduction in Manufacturing: The elimination of stoichiometric transition metals significantly lowers the bill of materials, while the simplified purification process reduces solvent usage and waste disposal costs. The use of inexpensive copper chloride as a catalyst compared to precious metals like palladium or rhodium offers immediate savings on raw material expenditures. Additionally, the high functional group tolerance minimizes the need for protecting groups, shortening the overall synthetic sequence and reducing the cumulative yield losses associated with multi-step processes. These factors combine to create a leaner, more cost-effective manufacturing profile that enhances the competitiveness of the final API.
- Enhanced Supply Chain Reliability: The reliance on commercially available and stable starting materials such as enaminones and hydrazones ensures a robust supply chain that is less susceptible to disruptions. Unlike methods requiring exotic or custom-synthesized reagents with long lead times, the inputs for this process are accessible from multiple global suppliers. The operational robustness of the reaction, characterized by its tolerance to varying conditions and substrates, translates to higher batch-to-batch consistency. This reliability is essential for maintaining continuous production schedules and meeting the stringent delivery timelines required by pharmaceutical clients.
- Scalability and Environmental Compliance: The process is inherently scalable, having been demonstrated to work effectively with various substrates under consistent conditions. The reduction in metal waste aligns with increasingly stringent environmental regulations regarding heavy metal discharge in pharmaceutical manufacturing. By generating less hazardous waste and utilizing milder conditions, the process supports sustainability goals and simplifies the regulatory approval process for new drug applications. The ability to produce high-purity intermediates with minimal environmental impact positions this technology as a future-proof solution for green chemistry initiatives.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this trifluoromethyl dihydrofuran synthesis technology. These insights are derived directly from the experimental data and scope defined in the patent literature, providing a clear understanding of the method's capabilities and limitations for potential partners and licensees.
Q: What are the advantages of this Cu-catalyzed method over traditional dihydrofuran synthesis?
A: Unlike conventional methods that often require stoichiometric transition metals and harsh conditions, this patent describes a catalytic system using only 5 mol% CuCl2 at mild temperatures (80-100°C), significantly improving functional group tolerance and reducing metal waste.
Q: Can this method construct quaternary carbon stereocenters effectively?
A: Yes, the reaction is specifically designed to generate stereoselective trifluoromethyl-substituted dihydrofuran derivatives containing a quaternary carbon stereocenter, which is a challenging structural motif in medicinal chemistry.
Q: Is the resulting dihydrofuran amine stable for further derivatization?
A: The synthesized dihydrofuran amines serve as robust synthetic intermediates. They can be quantitatively converted into trifluoromethyl-containing 1,4-dicarbonyl compounds via acid hydrolysis, expanding their utility in heterocycle construction.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Trifluoromethyl Dihydrofuran Amine Supplier
At NINGBO INNO PHARMCHEM, we recognize the strategic importance of accessing advanced synthetic methodologies to accelerate drug development timelines. Our team of expert process chemists has extensively evaluated the technology described in CN110922369B and is fully prepared to translate this laboratory-scale innovation into commercial reality. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project can seamlessly transition from clinical trials to market launch. Our state-of-the-art facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications, guaranteeing that every batch of trifluoromethyl-substituted dihydrofuran amine meets the highest quality standards required by global regulatory bodies.
We invite you to collaborate with us to leverage this cutting-edge chemistry for your next project. By partnering with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific molecule, demonstrating exactly how this route can optimize your budget. We encourage you to reach out today to discuss your requirements, obtain specific COA data for similar structures, and receive comprehensive route feasibility assessments that will de-risk your supply chain and secure your production future.