Advanced Catalytic Strategy for Stereoselective Trifluoromethyl Dihydrofuran Amines Manufacturing
Advanced Catalytic Strategy for Stereoselective Trifluoromethyl Dihydrofuran Amines Manufacturing
The rapid evolution of medicinal chemistry has placed a premium on fluorinated heterocyclic scaffolds, particularly those capable of modulating metabolic stability and lipophilicity in drug candidates. Patent CN110922369B introduces a groundbreaking methodology for the construction of trifluoromethyl-substituted dihydrofuran amine compounds, addressing a critical gap in the synthesis of complex fluorinated architectures. This technology leverages a copper-catalyzed cascade reaction to efficiently assemble these valuable motifs from readily available acyclic precursors. By enabling the direct formation of a quaternary carbon stereocenter within the dihydrofuran ring, this process offers a streamlined route to high-value pharmaceutical intermediates that were previously difficult to access. The strategic incorporation of the trifluoromethyl group at the C3 position not only enhances the biological profile of the resulting molecules but also provides a robust handle for further derivatization, making this patent a cornerstone for modern API development.
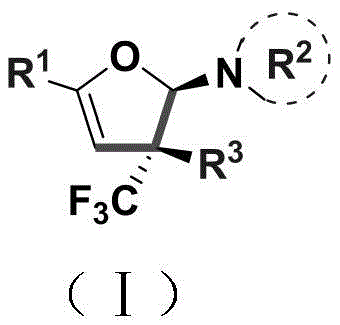
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of 2,3-dihydrofuran rings has relied heavily on classical cycloaddition strategies such as [4+1] or [3+2] annulations, which often suffer from significant practical drawbacks in an industrial setting. Traditional protocols frequently necessitate the use of stoichiometric amounts of expensive or toxic transition metals, leading to substantial waste generation and complicating downstream purification processes required for GMP compliance. Furthermore, many existing methods operate under extremely harsh reaction conditions, involving high temperatures or strong bases that limit functional group tolerance, thereby restricting the diversity of substrates that can be utilized. The difficulty in introducing a trifluoromethyl group directly during the ring-closing step has been a persistent bottleneck, often requiring multi-step sequences that erode overall yield and increase production costs. These inefficiencies create a pressing need for a more atom-economical and operationally simple approach to accessing these privileged scaffolds.
The Novel Approach
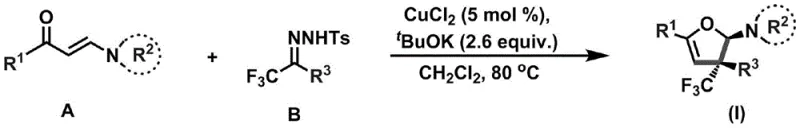
The methodology disclosed in CN110922369B represents a paradigm shift by utilizing a copper-catalyzed coupling of enaminones and trifluoromethylhydrazones to construct the dihydrofuran core in a single pot. As illustrated in the reaction scheme below, this approach merges the formation of the C-C and C-O bonds simultaneously, driven by a catalytic system comprising copper chloride and potassium tert-butoxide. This novel pathway eliminates the need for pre-functionalized cyclic starting materials, allowing chemists to build complexity from simple linear precursors with high stereoselectivity. The reaction proceeds under relatively mild thermal conditions (80-100°C) in common solvents like dichloromethane, significantly reducing energy consumption and safety risks associated with high-pressure or cryogenic reactions. By achieving the installation of the trifluoromethyl group and the heterocyclic ring in one concerted operation, this method drastically simplifies the synthetic route, offering a compelling solution for cost reduction in pharmaceutical intermediate manufacturing.
Mechanistic Insights into Cu-Catalyzed Cyclization
The success of this transformation hinges on the unique reactivity of the copper catalyst in mediating the interaction between the electron-rich enaminone and the electrophilic trifluoromethylhydrazone species. Mechanistically, the base (tBuOK) likely facilitates the deprotonation of the hydrazone to generate a reactive diazo or carbenoid intermediate in situ, which is then intercepted by the copper center. This metal-carbene species undergoes a selective insertion or cyclopropanation-like event with the enaminone double bond, followed by a rearrangement that establishes the dihydrofuran ring system. The presence of the bulky trifluoromethyl group adjacent to the reaction center plays a crucial role in directing the stereochemical outcome, ensuring the formation of the desired quaternary stereocenter with high fidelity. Understanding this catalytic cycle is vital for process chemists aiming to optimize reaction parameters for scale-up, as subtle changes in ligand environment or base strength could influence the ratio of diastereomers formed.
From an impurity control perspective, the mildness of the reaction conditions contributes to a cleaner crude profile, minimizing the formation of polymeric byproducts often seen in aggressive radical cyclizations. The use of a heterogeneous workup involving simple filtration to remove insoluble copper salts and base residues streamlines the isolation process, reducing the burden on chromatographic purification steps. This is particularly advantageous when scaling to multi-kilogram batches, where efficient removal of metal contaminants is a regulatory requirement. Furthermore, the tolerance of the catalytic system towards various electronic properties on the aryl rings of both substrates suggests a robust mechanism that is not easily poisoned by common functional groups. This resilience ensures consistent batch-to-batch quality, a critical factor for maintaining supply chain integrity in the production of high-purity pharmaceutical intermediates.
How to Synthesize Trifluoromethyl Dihydrofuran Amine Efficiently
The practical implementation of this synthesis involves a straightforward procedure that balances reaction efficiency with ease of operation, making it suitable for both laboratory discovery and pilot plant production. The process begins with the precise mixing of the enaminone and trifluoromethylhydrazone substrates in a defined molar ratio, typically favoring a slight excess of the hydrazone to drive the equilibrium forward. Following the addition of the copper catalyst and base under an inert atmosphere, the reaction mixture is heated to promote the cyclization event over a period of 48 to 72 hours. Detailed standard operating procedures regarding specific stoichiometry, solvent volumes, and purification gradients are essential for reproducibility and are outlined in the technical guide below.
- Mix enaminone (Compound A), trifluoromethylhydrazone (Compound B), copper chloride catalyst, and potassium tert-butoxide under inert gas protection.
- Add dichloromethane solvent and heat the reaction mixture to 80-100°C, stirring continuously for 48-72 hours to ensure complete cyclization.
- Filter off insoluble solids, concentrate the filtrate under reduced pressure, and purify the crude residue via column chromatography to isolate the target product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement specialists and supply chain managers, the adoption of this patented technology offers tangible benefits that extend beyond mere chemical novelty, directly impacting the bottom line through process intensification. The elimination of stoichiometric heavy metal reagents significantly reduces the cost of goods sold (COGS) by lowering raw material expenses and simplifying waste disposal protocols. Moreover, the ability to synthesize these complex scaffolds from commodity chemicals enhances supply security, as the reliance on exotic or hard-to-source starting materials is minimized. The robustness of the reaction conditions allows for greater flexibility in manufacturing scheduling, reducing the risk of batch failures due to sensitive parameters. This reliability translates into shorter lead times and a more resilient supply chain for critical drug substances.
- Cost Reduction in Manufacturing: The transition from stoichiometric to catalytic metal usage represents a major economic advantage, as copper chloride is inexpensive compared to precious metals like palladium or rhodium often used in similar transformations. Additionally, the simplified workup procedure, which relies on filtration and concentration rather than complex extractions or scavenging treatments, reduces solvent consumption and labor hours. These factors collectively contribute to a leaner manufacturing process that maximizes resource efficiency while maintaining high product quality standards.
- Enhanced Supply Chain Reliability: The broad substrate scope demonstrated in the patent indicates that the process is not limited to a narrow set of proprietary building blocks, allowing for the sourcing of diverse R1 and R3 variants from multiple global suppliers. This flexibility mitigates the risk of supply disruptions caused by single-source dependencies. Furthermore, the stability of the intermediates and the final dihydrofuran products ensures that inventory can be managed effectively without significant degradation, supporting just-in-time delivery models for downstream API synthesis.
- Scalability and Environmental Compliance: The use of dichloromethane as a solvent, while common, can be optimized or substituted in larger scale operations to meet evolving environmental regulations, and the low catalyst loading minimizes the heavy metal load in wastewater. The reaction's ability to proceed at moderate temperatures (80-100°C) reduces the energy footprint of the manufacturing process compared to high-temperature pyrolysis or cryogenic methods. These attributes facilitate the commercial scale-up of complex pharmaceutical intermediates while adhering to strict green chemistry principles and regulatory guidelines.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these trifluoromethylated heterocycles. The answers are derived directly from the experimental data and scope limitations defined in the patent literature, providing a realistic assessment of the technology's capabilities. Understanding these nuances helps stakeholders make informed decisions about integrating this chemistry into their development pipelines.
Q: What are the key advantages of this Cu-catalyzed method over traditional cycloadditions?
A: Unlike conventional methods that often require stoichiometric transition metals or harsh conditions, this protocol utilizes a catalytic amount of copper chloride under mild thermal conditions (80-100°C), offering superior functional group tolerance and operational simplicity.
Q: Can this method accommodate diverse substrate scopes for drug discovery?
A: Yes, the methodology demonstrates excellent substrate applicability, tolerating various R1 groups including alkyl, aryl, and heterocycles, as well as diverse R3 aryl substituents, making it highly valuable for generating libraries of fluorinated heterocycles.
Q: Is the resulting dihydrofuran amine stable for downstream processing?
A: The synthesized compounds possess a quaternary carbon stereocenter and can be quantitatively converted into trifluoromethyl-containing 1,4-dicarbonyl compounds under acidic conditions, serving as versatile intermediates for further heterocyclic construction.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Trifluoromethyl Dihydrofuran Amine Supplier
At NINGBO INNO PHARMCHEM, we recognize the strategic importance of fluorinated heterocycles in next-generation therapeutics and have invested heavily in mastering the complex chemistry required to produce them. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet the rigorous demands of global pharmaceutical clients. We adhere to stringent purity specifications and utilize rigorous QC labs to guarantee that every batch of trifluoromethyl-substituted dihydrofuran amine meets the highest industry standards for impurity profiles and stereochemical integrity.
We invite you to collaborate with us to leverage this innovative synthetic route for your specific drug development projects. Our technical sales team is ready to provide a Customized Cost-Saving Analysis tailored to your volume requirements, demonstrating how our optimized processes can reduce your overall procurement costs. Please contact our technical procurement team today to request specific COA data and route feasibility assessments for your target molecules, and let us support your journey from discovery to commercialization.