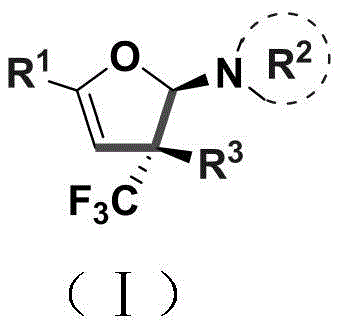
Scalable Synthesis of Trifluoromethyl Dihydrofuran Amines for Advanced Drug Discovery
Introduction to Advanced Trifluoromethyl Heterocycle Synthesis
The integration of trifluoromethyl groups into heterocyclic scaffolds represents a critical frontier in modern medicinal chemistry, driven by the unique metabolic stability and lipophilicity these motifs impart to drug candidates. Patent CN110922369B introduces a groundbreaking methodology for the construction of trifluoromethyl-substituted dihydrofuran amines, addressing long-standing challenges in accessing these complex molecular architectures. This technology leverages a copper-catalyzed cascade reaction that efficiently builds the five-membered dihydrofuran ring while simultaneously installing a quaternary carbon stereocenter, a structural feature highly prized for enhancing the three-dimensional complexity and binding affinity of pharmaceutical agents. The significance of this invention lies not only in its chemical elegance but also in its potential to streamline the supply chain for high-value intermediates used in the development of next-generation therapeutics and agrochemicals.
Traditionally, the synthesis of such densely functionalized heterocycles has been plagued by multi-step sequences, low atom economy, and the requirement for expensive precious metal catalysts. The approach detailed in this patent circumvents these bottlenecks by utilizing readily available acyclic precursors—specifically enaminones and trifluoromethylhydrazones—to achieve direct cyclization. This strategic shift from complex pre-functionalized cyclic starting materials to simple linear building blocks drastically reduces the cumulative cost of goods and minimizes waste generation. For R&D teams focused on rapid analog synthesis, the ability to access a diverse library of dihydrofuran derivatives through simple variation of the R1 and R3 substituents offers a powerful tool for structure-activity relationship (SAR) studies without compromising on purity or stereochemical integrity.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of 2,3-dihydrofuran rings, particularly those bearing trifluoromethyl groups at the C3 position, has relied on methodologies that are often operationally cumbersome and economically inefficient. Classical approaches frequently involve [4+1] or [3+2] cycloadditions that require stoichiometric amounts of transition metals, such as silver or palladium, which not only drive up raw material costs but also introduce significant challenges in downstream purification due to heavy metal residues. Furthermore, many existing protocols necessitate harsh reaction conditions, including extreme temperatures or strongly acidic/basic environments, which can lead to the decomposition of sensitive functional groups and limit the scope of compatible substrates. These limitations severely restrict the utility of conventional methods in the context of late-stage functionalization, where the preservation of delicate molecular features is paramount for maintaining biological activity.
Another critical drawback of prior art methods is the difficulty in controlling stereoselectivity, particularly when generating quaternary carbon centers. The formation of all-carbon quaternary stereocenters is notoriously difficult due to steric congestion, often resulting in poor diastereomeric ratios or the formation of racemic mixtures that require costly chiral resolution steps. Additionally, the reliance on pre-synthesized cyclic precursors or specialized reagents increases the lead time for project initiation, creating bottlenecks in the drug discovery pipeline. The inability to efficiently introduce the trifluoromethyl group in a one-pot fashion alongside ring closure further complicates the synthetic route, often necessitating separate fluorination steps that utilize hazardous reagents like Selectfluor or DAST, thereby raising safety and environmental concerns for large-scale manufacturing operations.
The Novel Approach
In stark contrast to these legacy techniques, the novel catalytic system disclosed in the patent employs a robust copper chloride catalyst in conjunction with potassium tert-butoxide to facilitate a smooth and selective cyclization. This method operates under relatively mild thermal conditions, typically between 80°C and 100°C, which significantly lowers energy consumption and enhances process safety compared to high-temperature alternatives. The use of dichloromethane as a solvent provides an optimal medium for solubilizing both the organic substrates and the inorganic base, ensuring homogeneous reaction kinetics that contribute to consistent batch-to-batch reproducibility. By utilizing a catalytic amount of copper (approximately 5 mol%), the process achieves high turnover numbers, effectively decoupling the cost of the catalyst from the scale of production and making it economically viable for kilogram-to-ton manufacturing campaigns.
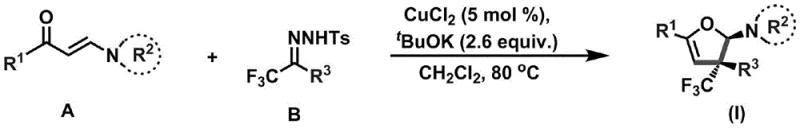
The core innovation of this approach is the tandem nature of the reaction, which combines C-N bond formation, C-C bond formation, and ring closure into a single operational step. This convergence not only improves the overall yield by minimizing intermediate isolation losses but also dramatically simplifies the workflow for process chemists. The reaction exhibits exceptional functional group tolerance, successfully accommodating a wide array of substituents on both the enaminone and the hydrazone components, including electron-rich and electron-deficient aryl groups, alkyl chains, and even heterocycles. This broad substrate scope means that a single standardized protocol can be applied to synthesize a vast library of analogs, accelerating the optimization phase of drug development. Moreover, the workup procedure is remarkably straightforward, involving simple filtration to remove inorganic salts followed by concentration and column chromatography, which facilitates rapid purification and high-purity isolation of the target dihydrofuran amines.
Mechanistic Insights into Cu-Catalyzed Cyclization
The mechanistic pathway of this transformation likely involves the activation of the trifluoromethylhydrazone by the base to generate a diazo species or a metal-carbene intermediate, which subsequently undergoes insertion or addition reactions with the electron-rich enaminone double bond. The copper catalyst plays a pivotal role in mediating this reactivity, potentially stabilizing reactive intermediates and lowering the activation energy barrier for the cyclization step. The presence of the trifluoromethyl group adjacent to the reaction center exerts a strong electronic influence, which, when coupled with the steric bulk of the substituents, directs the formation of the quaternary stereocenter with high selectivity. Understanding this mechanism is crucial for process optimization, as it highlights the importance of maintaining strict anhydrous conditions and precise stoichiometric control of the base to prevent premature decomposition of the diazo intermediate or side reactions such as dimerization.
From an impurity control perspective, the mildness of the reaction conditions inherently suppresses the formation of thermal degradation products that are common in harsher cyclization protocols. The high chemoselectivity of the copper catalyst ensures that other sensitive functionalities present on the substrate, such as halides or esters, remain intact, thereby preserving the integrity of the molecular scaffold for subsequent derivatization. The stereoselective outcome is particularly valuable, as the resulting dihydrofuran amines possess defined spatial arrangements that are essential for specific receptor interactions in biological systems. This level of control eliminates the need for extensive chiral separation processes, which are often the most cost-prohibitive step in the manufacture of chiral intermediates. Consequently, the process delivers a product with a clean impurity profile, reducing the burden on analytical quality control and ensuring compliance with stringent regulatory standards for pharmaceutical ingredients.
How to Synthesize Trifluoromethyl Dihydrofuran Amine Efficiently
The practical implementation of this synthesis is designed for scalability and ease of execution in standard laboratory or pilot plant settings. The protocol begins with the preparation of the requisite starting materials, enaminone (Compound A) and trifluoromethylhydrazone (Compound B), which can be synthesized via established literature procedures or sourced commercially. The reaction is initiated by combining these precursors with the copper catalyst and base under an inert atmosphere to prevent oxidation of sensitive intermediates. Heating the mixture to the specified temperature range drives the reaction to completion over a period of 48 to 72 hours, allowing sufficient time for the thermodynamic equilibration towards the desired stereoisomer. Following the reaction, the mixture is cooled and filtered to remove insoluble inorganic byproducts, such as potassium chloride and copper salts, yielding a clear solution that can be directly concentrated. The crude product is then subjected to flash column chromatography using a petroleum ether and ethyl acetate gradient to isolate the pure dihydrofuran amine derivative.
- Mix compound A (enaminone), compound B (trifluoromethylhydrazone), copper chloride (5 mol%), and potassium tert-butoxide (2.6 equiv.) under inert gas protection.
- Add dichloromethane solvent and heat the reaction solution to 80-100°C, stirring continuously for 48-72 hours to ensure complete cyclization.
- Filter off insoluble solids, concentrate the filtrate under reduced pressure, and purify the residue via column chromatography to obtain the target dihydrofuran amine.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this patented technology offers substantial strategic benefits that extend beyond mere chemical novelty. The primary advantage lies in the significant reduction of raw material costs achieved by replacing expensive stoichiometric reagents with catalytic quantities of commodity chemicals like copper chloride. This shift fundamentally alters the cost structure of the synthesis, making the final intermediate more price-competitive in the global market. Furthermore, the use of simple, non-proprietary starting materials mitigates supply risk, as these commodities are widely available from multiple vendors, ensuring business continuity and preventing bottlenecks caused by single-source dependencies. The robustness of the reaction conditions also implies a lower rate of batch failures, leading to more predictable production schedules and reliable delivery timelines for downstream customers.
- Cost Reduction in Manufacturing: The elimination of precious metal catalysts and the simplification of the synthetic route from multi-step to one-pot directly translates to lower operational expenditures. By removing the need for specialized equipment capable of withstanding extreme pressures or temperatures, capital investment requirements are minimized. Additionally, the simplified workup procedure reduces solvent consumption and waste disposal costs, contributing to a leaner and more sustainable manufacturing process that aligns with modern green chemistry principles and corporate sustainability goals.
- Enhanced Supply Chain Reliability: The broad substrate scope of this methodology allows for the flexible sourcing of diverse building blocks, enabling manufacturers to quickly adapt to fluctuations in the availability or pricing of specific raw materials. The high yield and selectivity of the reaction ensure that less starting material is wasted, maximizing the throughput of existing production facilities. This efficiency gain effectively increases capacity without the need for physical expansion, allowing suppliers to meet surging demand for complex heterocyclic intermediates with greater agility and responsiveness to market dynamics.
- Scalability and Environmental Compliance: The mild reaction conditions and the absence of hazardous fluorinating agents make this process inherently safer and easier to scale from gram to ton quantities. The reduced generation of toxic waste streams simplifies environmental compliance and lowers the cost associated with effluent treatment. This environmental friendliness is increasingly becoming a key differentiator in supplier selection, as pharmaceutical companies prioritize partners who can demonstrate a commitment to reducing their carbon footprint and adhering to strict environmental, social, and governance (ESG) criteria.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation and application of this trifluoromethyl dihydrofuran synthesis technology. These insights are derived directly from the experimental data and scope analysis presented in the patent documentation, providing a realistic overview of what potential partners and licensees can expect. Understanding these details is essential for evaluating the feasibility of integrating this chemistry into existing production pipelines or research programs.
Q: What are the key advantages of this Cu-catalyzed cyclization method?
A: The method utilizes mild reaction conditions (80-100°C) and catalytic amounts of inexpensive copper chloride, avoiding the need for stoichiometric transition metals or harsh reagents often required in traditional dihydrofuran synthesis.
Q: Can this process tolerate diverse functional groups on the substrate?
A: Yes, the patent demonstrates excellent substrate applicability, successfully accommodating various R1 groups including alkyl, aryl, heterocycles, and ferrocene, as well as diverse R3 aryl substituents with electron-withdrawing or donating groups.
Q: How can the resulting dihydrofuran amine be further derivatized?
A: The synthesized dihydrofuran amine serves as a versatile intermediate that can be quantitatively converted into trifluoromethyl-containing 1,4-dicarbonyl compounds via acid hydrolysis using TsOH·H2O at room temperature.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Trifluoromethyl Dihydrofuran Amine Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of this copper-catalyzed cyclization technology in advancing the development of novel therapeutic agents. As a leading CDMO partner, we possess the technical expertise and infrastructure necessary to translate this academic innovation into a robust commercial reality. Our team has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project can seamlessly transition from bench-scale discovery to full-scale manufacturing. We are committed to delivering high-purity intermediates that meet stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation to verify identity, potency, and impurity profiles.
We invite you to collaborate with us to leverage this cutting-edge synthesis for your next drug discovery program. By partnering with NINGBO INNO PHARMCHEM, you gain access to a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality needs. We encourage you to contact our technical procurement team today to request specific COA data for our catalog compounds or to discuss custom route feasibility assessments for your proprietary targets. Let us help you accelerate your timeline to market with a reliable, cost-effective, and scalable supply of these critical trifluoromethyl-substituted building blocks.