Advanced Decitabine Manufacturing: Overcoming Isomer Separation Challenges for Commercial Scale
The pharmaceutical industry continuously seeks robust synthetic pathways for oncology therapeutics, particularly for nucleoside analogues like Decitabine, which play a pivotal role in treating myelodysplastic syndromes. A significant technological breakthrough in this domain is detailed in patent CN102010455A, which discloses a novel method for preparing Decitabine that fundamentally alters the economic and technical landscape of its production. This patent introduces a streamlined synthesis strategy that begins with 4-amino-1-(β-D-erythro ribofuranose)-1,3,5-triazine-2(1H)-ketone, a strategic starting material that inherently possesses the required stereochemistry. By leveraging this specific precursor, the invention successfully circumvents the historically problematic and yield-limiting step of separating alpha and beta isomers, a bottleneck that has plagued conventional manufacturing processes for decades. The result is a concise, four-step synthetic sequence capable of delivering Decitabine with a purity exceeding 99.85% under mild reaction conditions, representing a substantial advancement for reliable API intermediate suppliers aiming to optimize their production portfolios.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthetic routes for Decitabine typically commence with 2-deoxy-D-ribose or similar sugar derivatives, which inevitably leads to the formation of a mixture of anomeric isomers during the glycosylation or condensation phases. In these legacy processes, the reaction between the sugar moiety and the heterocyclic base generates both the desired beta-anomer (Decitabine) and the inactive alpha-anomer in significant quantities. Separating these stereoisomers is notoriously difficult and resource-intensive, often requiring repeated recrystallizations or extensive column chromatography, which drastically reduces the overall yield and increases the cost of goods sold. Furthermore, the use of protecting groups like methoxyacetic anhydride in some prior art methods complicates the deprotection stages, leading to wayward reaction processes and loaded down with trivial details that hinder industrial scalability. The inability to efficiently split these isomers not only impacts the final purity but also creates significant waste streams, posing challenges for environmental compliance and supply chain efficiency in large-scale manufacturing facilities.
The Novel Approach
In stark contrast to these cumbersome legacy techniques, the methodology disclosed in patent CN102010455A adopts a retrosynthetic logic that prioritizes stereochemical integrity from the outset. By selecting 4-amino-1-(β-D-erythro ribofuranose)-1,3,5-triazine-2(1H)-ketone as the raw material, the synthesis effectively locks the stereochemistry at the anomeric center before the critical deoxygenation step occurs. This strategic choice eliminates the generation of the unwanted alpha-isomer entirely, thereby removing the need for complex isomer separation protocols. The process involves a logical sequence of etherification, condensation, deprotection, and specifically, the stripping of the 2-position hydroxyl group. This approach not only simplifies the operational workflow but also ensures that the final product is obtained with exceptional purity directly from the reaction mixture. The elimination of the isomer splitting step represents a paradigm shift in cost reduction in API intermediate manufacturing, allowing producers to achieve higher throughput with fewer unit operations and reduced solvent consumption.
Mechanistic Insights into Radical Deoxygenation and Stereocontrol
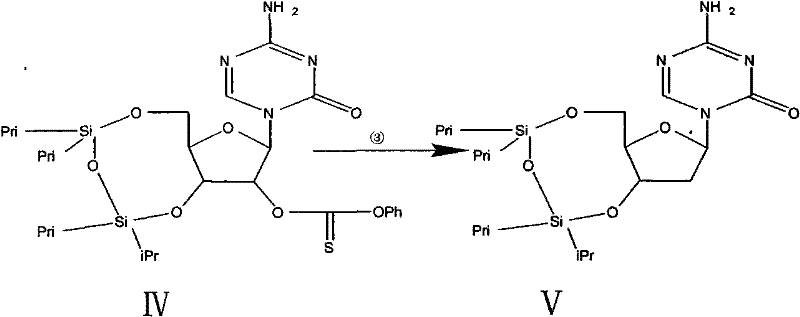
The core chemical transformation in this novel pathway is the selective deoxygenation at the C-2 position of the ribofuranose ring, a step that converts the ribose derivative into the requisite 2'-deoxy structure of Decitabine. This transformation is achieved through a Barton-McCombie type deoxygenation mechanism, which is renowned for its ability to replace hydroxyl groups with hydrogen atoms under mild conditions. Initially, the 2-hydroxyl group of the protected intermediate is activated by conversion into a thiocarbonate derivative using phenoxy thiocarbonyl chloride and a catalytic amount of DMAP. This activation is crucial as it transforms a poor leaving group (hydroxyl) into a species susceptible to radical attack. Subsequently, the thiocarbonate intermediate undergoes homolytic cleavage initiated by a radical source, typically AIBN (azobisisobutyronitrile), in the presence of a hydrogen donor like tributyltin hydride. The resulting carbon-centered radical at the C-2 position abstracts a hydrogen atom from the tin hydride, completing the deoxygenation while preserving the stereochemical configuration at the adjacent anomeric center.
From an impurity control perspective, this radical mechanism offers distinct advantages over acid-catalyzed dehydration or other ionic methods that might risk anomerization or degradation of the sensitive triazine base. The use of mild temperatures during the initial protection steps (controlled between -10°C and 20°C) further minimizes the formation of thermal degradation byproducts. Moreover, the final deprotection step utilizing tetrabutylammonium fluoride (TBAF) is highly selective for silyl ethers, ensuring that the glycosidic bond remains intact while removing the bulky TIPDS protecting group. This precision in reaction engineering ensures that the impurity profile of the final Decitabine is exceptionally clean, meeting the stringent requirements for high-purity pharmaceutical intermediates. The structural integrity of the final product is confirmed through rigorous analysis, including X-ray diffraction, which validates the beta-configuration essential for biological activity.
How to Synthesize Decitabine Efficiently
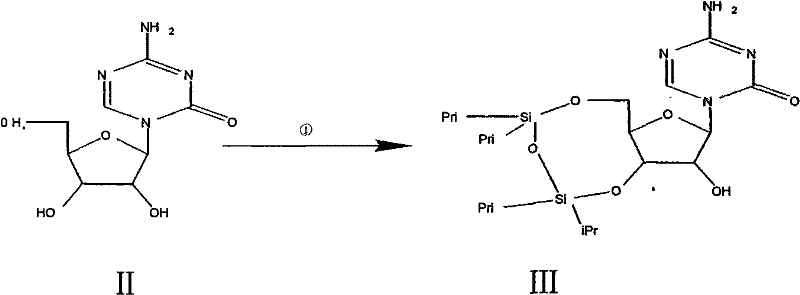
The synthesis of Decitabine via this patented route is characterized by its operational simplicity and high reproducibility, making it an ideal candidate for technology transfer and commercial adoption. The process begins with the protection of the 3,5-hydroxyl groups of the starting nucleoside using 1,3-dichloro-1,1,3,3-tetraisopropyldisiloxane (TIPDS-Cl2) in anhydrous pyridine, forming a bridged siloxane ring that stabilizes the sugar conformation. Following isolation of this protected intermediate, the 2-hydroxyl group is activated and subsequently removed via radical chemistry, as detailed in the mechanistic section. The final step involves the removal of the silyl protecting groups to reveal the free hydroxyls of the Decitabine molecule. For research and development teams looking to implement this pathway, the detailed standardized synthetic steps are provided in the guide below, ensuring consistent results and high purity outcomes.
- Protect the 3,5-hydroxyl groups of the starting ribofuranose derivative using 1,3-dichloro-1,1,3,3-tetraisopropyldisiloxane (TIPDS-Cl2) in anhydrous pyridine to form Intermediate III.
- Activate the 2-position hydroxyl group of Intermediate III by reacting with phenoxy thiocarbonyl chloride and DMAP in acetonitrile to generate the thiocarbonate Intermediate IV.
- Perform radical deoxygenation using tributyltin hydride and AIBN in refluxing hexane to remove the 2-hydroxyl group, yielding the deoxy-intermediate V.
- Remove the silyl protecting groups using tetrabutylammonium fluoride (TBAF) in THF, followed by recrystallization in methanol to obtain pure Decitabine.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this synthesis method offers compelling strategic advantages that extend beyond mere technical elegance. The primary value driver is the significant simplification of the purification train, which directly correlates to reduced manufacturing costs and improved supply reliability. By eliminating the need for isomer separation, manufacturers can drastically reduce the volume of solvents and stationary phases required, leading to substantial cost savings in raw materials and waste disposal. Furthermore, the shorter synthetic sequence reduces the overall cycle time, allowing for faster turnaround from raw material intake to finished goods, which is critical for maintaining inventory levels in a volatile market. The robustness of the reaction conditions, which avoid extreme temperatures or hazardous high-pressure environments, also enhances workplace safety and reduces the capital expenditure required for specialized reactor infrastructure.
- Cost Reduction in Manufacturing: The most significant economic benefit of this process lies in the avoidance of isomer separation. In traditional routes, separating alpha and beta anomers often consumes more than half of the potential yield and requires expensive chromatographic resources. By starting with a stereochemically pure precursor, this method effectively doubles the theoretical yield relative to non-stereoselective routes, leading to a drastic reduction in the cost per kilogram of the active pharmaceutical ingredient. Additionally, the use of common reagents like TIPDS-Cl2 and tributyltin hydride, which are readily available in the global chemical market, ensures that raw material costs remain stable and predictable, shielding the supply chain from price volatility associated with exotic catalysts.
- Enhanced Supply Chain Reliability: Supply continuity is paramount for oncology drugs, where patient treatment schedules cannot be interrupted. This synthesis route enhances reliability by reducing the number of critical process parameters that could lead to batch failure. The elimination of the unpredictable isomer splitting step removes a major source of yield variability, ensuring that every batch meets the strict purity specifications of >99.85%. Furthermore, the intermediates generated in this pathway, such as the silyl-protected derivatives, are generally stable and can be stored or transported if necessary, providing flexibility in production scheduling and inventory management for global distribution networks.
- Scalability and Environmental Compliance: As regulatory bodies impose stricter limits on heavy metal residues and solvent emissions, this process demonstrates strong alignment with green chemistry principles. Although tributyltin hydride is used, it is consumed in a stoichiometric manner and can be managed through established waste treatment protocols, avoiding the persistent contamination risks associated with transition metal catalysts like palladium or platinum. The mild reaction temperatures and the ability to perform reactions in standard solvents like acetonitrile and hexane facilitate easy scale-up from pilot plant to multi-ton commercial production without the need for complex engineering modifications, ensuring a seamless transition from R&D to full-scale manufacturing.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this Decitabine synthesis technology. These insights are derived directly from the experimental data and technical specifications outlined in the patent documentation, providing a clear understanding of the process capabilities and limitations for potential partners and licensees.
Q: How does this synthesis method avoid the difficult separation of alpha and beta isomers?
A: Unlike traditional methods that start with 2-deoxy-D-ribose and generate a mixture of anomers requiring complex separation, this patent utilizes 4-amino-1-(β-D-erythro ribofuranose)-1,3,5-triazine-2(1H)-ketone as the starting material. Since the beta-configuration is already established in the starting material, the subsequent deoxygenation at the 2-position proceeds with retention of configuration, effectively bypassing the need for isomer splitting entirely.
Q: What is the achieved purity of Decitabine using this specific protocol?
A: The patent data explicitly states that the final product obtained through this concise synthesis process achieves a purity of greater than 99.85% as detected by HPLC. This high level of purity is critical for pharmaceutical applications to minimize impurity-related toxicity and ensure regulatory compliance.
Q: Is the radical deoxygenation step scalable for industrial production?
A: Yes, the process is designed for industrial applicability. The radical deoxygenation step utilizes standard reagents like tributyltin hydride and AIBN under reflux conditions in hexane. While tin removal requires attention, the overall route avoids cryogenic conditions and uses mild temperatures (-10°C to 20°C) for the protection steps, making it suitable for commercial scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Decitabine Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of high-quality intermediates in the development of life-saving cancer therapies. Our technical team has extensively analyzed the pathway described in patent CN102010455A and possesses the expertise to execute this sophisticated chemistry with precision. We bring extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with consistency and reliability. Our state-of-the-art facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications, guaranteeing that every batch of Decitabine or its intermediates meets the highest international standards for pharmaceutical use.
We invite you to collaborate with us to leverage this advanced synthesis technology for your product pipeline. By partnering with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage you to contact us today to discuss route feasibility assessments and to obtain specific COA data for our available intermediates, ensuring a secure and efficient supply chain for your Decitabine projects.