Advanced Synthesis of 5-Trifluoromethyl-1,2,4-Triazoles for Scalable Pharmaceutical Manufacturing
Advanced Synthesis of 5-Trifluoromethyl-1,2,4-Triazoles for Scalable Pharmaceutical Manufacturing
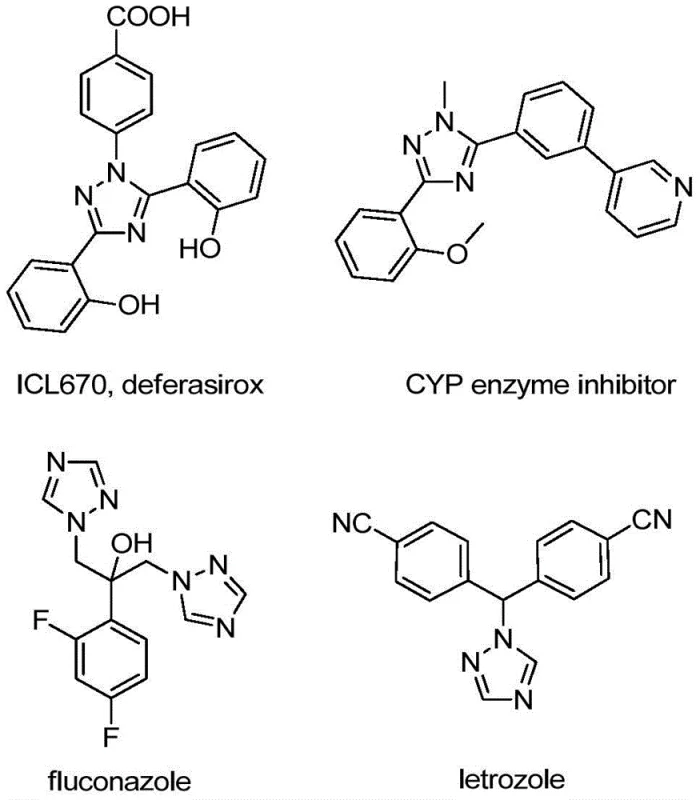
The pharmaceutical and agrochemical industries are constantly seeking robust, scalable, and cost-effective methodologies for constructing nitrogen-containing heterocycles, particularly those bearing trifluoromethyl groups which are renowned for enhancing metabolic stability and lipophilicity. Patent CN110467579B discloses a groundbreaking preparation method for 5-trifluoromethyl substituted 1,2,4-triazole compounds that addresses many of the historical pain points associated with this chemical class. This novel approach utilizes a non-metallic iodine-promoted cyclization strategy, starting from inexpensive and readily available hydrazones and trifluoroethylimidoyl chloride. Unlike conventional routes that often demand stringent anhydrous conditions or toxic heavy metal catalysts, this invention operates under relatively mild conditions, offering a streamlined pathway for the production of high-purity pharmaceutical intermediates. The significance of this technology lies not only in its chemical efficiency but also in its potential to drastically simplify supply chains for complex heterocyclic building blocks used in drug discovery and development.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of nitrogen-containing heterocycles bearing trifluoromethyl groups has been fraught with significant technical and economic challenges. Literature reports generally categorize existing methods into two main streams, both of which possess distinct drawbacks for large-scale manufacturing. The first approach involves the trifluoromethylation of pre-synthesized nitrogen-containing heterocycles, a process that typically necessitates the use of various specialized and often prohibitively expensive trifluoromethylating reagents. These reagents can be difficult to source in bulk and may pose handling hazards. The second mainstream method relies on reacting synthons bearing a trifluoromethyl group with suitable coupling substrates. While effective, the commonly used synthons such as trifluorodiazoethane are inherently unstable and potentially dangerous to handle on an industrial scale. Alternatively, trifluoroethylimide acid halides have been used but their application has not been widespread due to perceived limitations in reactivity or scope. Furthermore, many traditional protocols require rigorous exclusion of moisture and oxygen, demanding specialized equipment and increasing operational costs, while the reliance on transition metal catalysts introduces the risk of heavy metal contamination, necessitating costly purification steps to meet regulatory standards for active pharmaceutical ingredients.
The Novel Approach
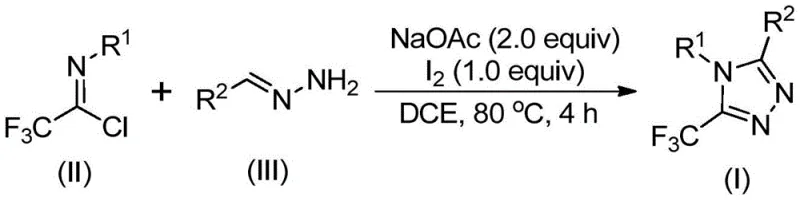
In stark contrast to these legacy methods, the technology described in patent CN110467579B introduces a simple, efficient, and economically superior route. By leveraging trifluoroethylimidoyl chloride and hydrazones as starting materials, the process bypasses the need for unstable diazo compounds or expensive late-stage trifluoromethylation agents. The core innovation lies in the use of elemental iodine as a promoter in conjunction with sodium acetate, facilitating a smooth cyclization without the need for toxic heavy metals. This method does not require anhydrous or anaerobic conditions, meaning reactions can be conducted in standard glassware without the need for inert gas manifolds or gloveboxes, significantly lowering the barrier to entry for production. The operational simplicity is matched by the versatility of the chemistry; through substrate design, variously substituted 1,2,4-triazole compounds with trifluoromethyl groups at different positions can be synthesized. This flexibility allows chemists to rapidly access diverse chemical space for structure-activity relationship (SAR) studies while maintaining a production-ready protocol that is convenient to operate and widens the applicability of the method across different therapeutic areas.
Mechanistic Insights into Iodine-Promoted Cyclization
The mechanistic pathway of this transformation is a sophisticated yet elegant sequence of bond-forming events driven by base promotion and oxidative iodination. The reaction is believed to initiate with a base-promoted intermolecular carbon-nitrogen bond formation between the trifluoroethylimidoyl chloride and the hydrazone, leading to the generation of a trifluoroacetamidine intermediate. Following this initial condensation, the system undergoes isomerization to align the reactive centers for cyclization. The introduction of elemental iodine then triggers a base-promoted oxidative iodination, generating a key iodinated intermediate. This species is primed for an intramolecular electrophilic substitution reaction, where the nucleophilic nitrogen attacks the electrophilic carbon, closing the five-membered ring. The final step involves aromatization, resulting in the stable 5-trifluoromethyl substituted 1,2,4-triazole core. Understanding this mechanism is crucial for process optimization, as it highlights the dual role of the reagents: sodium acetate acts as the base to drive condensation and deprotonation, while iodine serves as the oxidant to facilitate the final ring closure and aromatization.
From an impurity control perspective, this mechanism offers distinct advantages for ensuring high purity in the final product. Because the reaction proceeds through well-defined intermediates and avoids radical pathways often associated with harsh trifluoromethylation reagents, the formation of complex, hard-to-remove side products is minimized. The mild reaction conditions (80°C to 100°C) prevent thermal degradation of sensitive functional groups on the aryl or heteroaryl substituents. Moreover, the absence of transition metals eliminates the risk of metal-catalyzed side reactions or metal residue contamination, which is a critical quality attribute for pharmaceutical intermediates. The use of column chromatography as a standard purification technique, as suggested in the patent, indicates that the crude reaction mixtures are relatively clean, allowing for straightforward isolation of the target compound. This predictability in the reaction profile ensures consistent batch-to-batch quality, a paramount concern for supply chain reliability in the fine chemical sector.
How to Synthesize 5-Trifluoromethyl-1,2,4-Triazole Efficiently
Implementing this synthesis route in a laboratory or pilot plant setting requires adherence to specific stoichiometric ratios and thermal profiles to maximize yield and minimize waste. The patent outlines a robust protocol where sodium acetate, trifluoroethylimidoyl chloride, and hydrazone are combined in an organic solvent, preferably an aprotic solvent like dichloroethane (DCE), which effectively dissolves all reactants and promotes the reaction kinetics. The detailed standardized synthesis steps below provide a clear roadmap for executing this transformation, ensuring that the critical parameters regarding temperature, reaction time, and reagent addition order are strictly followed to achieve the reported high conversion rates.
- Combine sodium acetate, trifluoroethylimidoyl chloride, and hydrazone in an organic solvent such as dichloroethane (DCE) within a reaction vessel.
- Heat the mixture to a temperature range of 80°C to 100°C and maintain stirring for a duration of 2 to 4 hours to facilitate initial condensation.
- Introduce elemental iodine into the reaction system and continue heating for an additional 1 to 2 hours to promote oxidative cyclization and aromatization.
- Upon completion, perform post-treatment involving filtration, silica gel mixing, and purification via column chromatography to isolate the final triazole product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this synthetic methodology represents a strategic opportunity to optimize costs and enhance supply security for critical heterocyclic intermediates. The shift away from exotic reagents and complex reaction conditions translates directly into tangible operational efficiencies. By utilizing commodity chemicals as starting materials and avoiding the logistical burdens associated with hazardous or unstable reagents, manufacturers can streamline their inventory management and reduce the total cost of ownership for these valuable building blocks. The following points detail how this technology aligns with key commercial objectives in the fine chemical industry.
- Cost Reduction in Manufacturing: The economic benefits of this process are driven primarily by the elimination of expensive catalysts and reagents. Traditional methods often rely on precious metal catalysts or specialized trifluoromethylating agents that command high market prices and incur significant disposal costs. By replacing these with inexpensive elemental iodine and sodium acetate, the direct material costs are substantially reduced. Furthermore, the removal of heavy metals from the process flow eliminates the need for expensive scavenging resins or complex purification protocols designed to lower metal residues to ppm levels, thereby reducing downstream processing costs. The ability to run the reaction without strict anhydrous or anaerobic conditions also lowers utility costs associated with drying solvents and maintaining inert atmospheres, contributing to a leaner and more cost-effective manufacturing model.
- Enhanced Supply Chain Reliability: Supply chain resilience is heavily dependent on the availability and stability of raw materials. This method utilizes trifluoroethylimidoyl chloride and hydrazones, which are derived from widely available aromatic amines and aldehydes. These precursors are produced on a large scale globally, ensuring a stable and diversified supply base that mitigates the risk of shortages. Additionally, the stability of these starting materials allows for easier storage and transportation compared to sensitive reagents like trifluorodiazoethane. The robustness of the reaction conditions means that production is less susceptible to disruptions caused by equipment failure related to inert gas systems or moisture control, ensuring consistent delivery schedules and reliable fulfillment of customer orders for high-purity pharmaceutical intermediates.
- Scalability and Environmental Compliance: Scaling chemical processes from the bench to the plant floor often reveals hidden complexities, but this method is explicitly designed for scalability. The patent notes that the process can be easily expanded to the gram level and implies suitability for larger scales due to its operational simplicity. The use of common organic solvents like dichloroethane, which are standard in industrial settings, facilitates easy technology transfer. From an environmental standpoint, the avoidance of toxic heavy metals significantly reduces the burden of hazardous waste treatment and disposal. This aligns with increasingly stringent global environmental regulations and corporate sustainability goals. The simpler workup procedure, involving filtration and standard chromatography, minimizes solvent usage and waste generation compared to multi-step purification sequences, making the process not only commercially viable but also environmentally responsible.
Frequently Asked Questions (FAQ)
To further clarify the technical and commercial implications of this patent, we have compiled a set of frequently asked questions based on the detailed experimental data and background information provided in the documentation. These answers address common concerns regarding reaction conditions, substrate compatibility, and the practical advantages of this iodine-promoted cyclization over legacy methods. Understanding these nuances is essential for R&D teams evaluating this route for new project pipelines and for procurement specialists assessing the long-term viability of the supply chain.
Q: What are the primary advantages of this iodine-promoted method over traditional trifluoromethylation?
A: Unlike traditional methods that often require expensive trifluoromethylating reagents or unstable trifluorodiazoethane, this method utilizes cheap and readily available trifluoroethylimidoyl chloride and hydrazones. Furthermore, it eliminates the need for toxic heavy metal catalysts and strict anhydrous or anaerobic conditions, significantly simplifying operational complexity and reducing safety risks.
Q: Is this synthesis method suitable for large-scale industrial production?
A: Yes, the method is highly amenable to scale-up. The patent explicitly states that the process can be easily expanded to the gram level and beyond, providing a viable pathway for industrial mass production. The use of common organic solvents like dichloroethane and the tolerance for ambient atmospheric conditions further support its feasibility for commercial manufacturing environments.
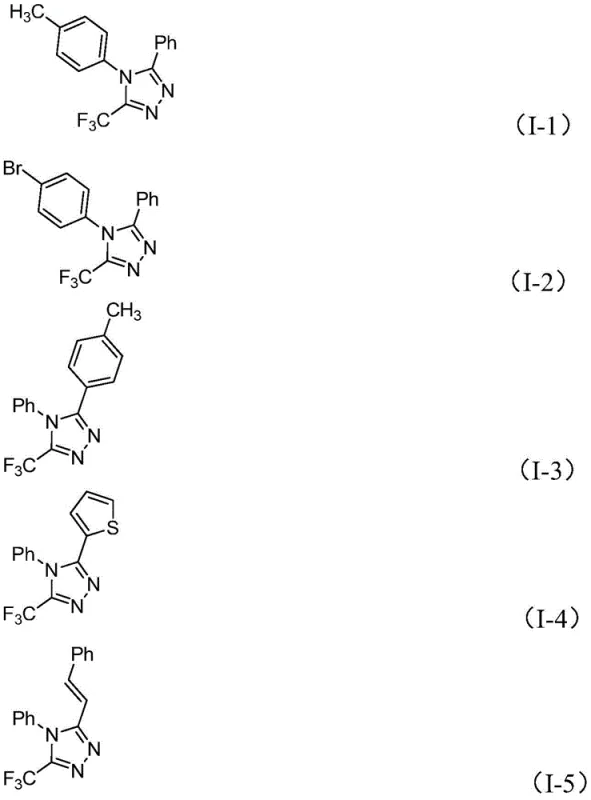
Q: What is the substrate scope for the R1 and R2 groups in this triazole synthesis?
A: The method demonstrates broad substrate tolerance. R1 can be a substituted or unsubstituted aryl group (e.g., phenyl with methyl, methoxy, bromo, or trifluoromethyl substituents), while R2 can be alkenyl, substituted or unsubstituted aryl, or heteroaryl groups. This versatility allows for the design and synthesis of various 1,2,4-triazole derivatives with different substitution patterns at the 4 and 5 positions.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 5-Trifluoromethyl-1,2,4-Triazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that advanced synthetic methodologies play in accelerating drug development and securing the supply of essential intermediates. Our team of expert chemists has thoroughly analyzed the potential of the iodine-promoted synthesis described in patent CN110467579B and is prepared to leverage this technology for your specific project needs. As a leading CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from laboratory discovery to industrial manufacturing is seamless. Our state-of-the-art facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications, guaranteeing that every batch of 5-trifluoromethyl-1,2,4-triazole delivered meets the highest quality standards required by the global pharmaceutical industry.
We invite you to collaborate with us to unlock the full potential of this efficient synthesis route. Whether you require custom synthesis of specific derivatives or large-scale production of the core scaffold, our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your volume requirements. We encourage you to contact us today to request specific COA data and route feasibility assessments, allowing us to demonstrate how our expertise can drive down your costs and secure your supply chain for these vital pharmaceutical intermediates.