Advanced Palladium-Catalyzed Carbonylation for Scalable Quinazolinone Pharmaceutical Intermediates
Advanced Palladium-Catalyzed Carbonylation for Scalable Quinazolinone Pharmaceutical Intermediates
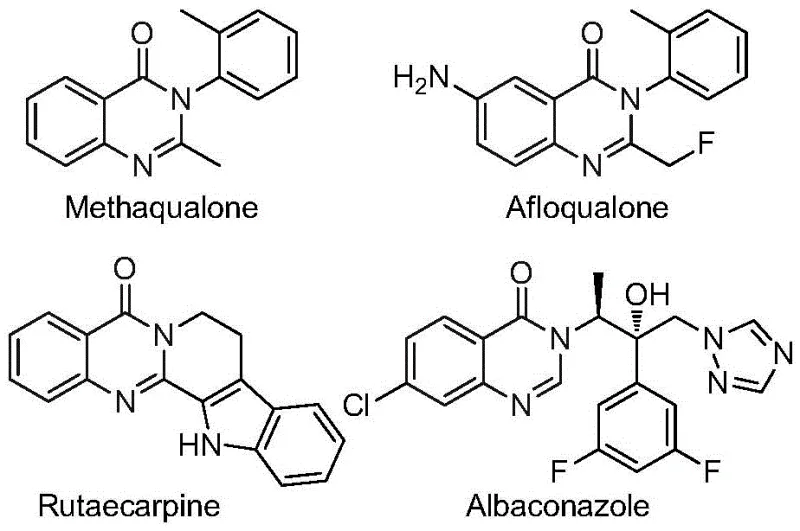
The quinazolinone scaffold represents a cornerstone structure in modern medicinal chemistry, underpinning a vast array of bioactive molecules with potent therapeutic profiles ranging from antifungal and antiviral activities to anticancer and anti-inflammatory properties. As depicted in the structural diversity of known drugs such as Methaqualone and Albaconazole, the incorporation of specific functional groups, particularly the trifluoromethyl moiety, can drastically enhance metabolic stability, lipophilicity, and bioavailability of the parent molecule. However, the efficient construction of 2-trifluoromethyl substituted quinazolinones has historically posed significant synthetic challenges due to the inertness of the C-F bonds and the difficulty in introducing the carbonyl group under mild conditions. Addressing these critical bottlenecks, the technology disclosed in patent CN112480015B introduces a transformative multi-component one-pot methodology that leverages transition metal catalysis to streamline the production of these high-value intermediates.
This innovative approach not only simplifies the operational workflow but also significantly broadens the substrate tolerance, allowing for the rapid generation of diverse chemical libraries essential for drug discovery campaigns. By utilizing readily available nitro compounds and trifluoroethylimidoyl chlorides as starting materials, the process circumvents the need for pre-activated substrates that often drive up costs and extend lead times in traditional synthetic routes. For R&D directors and process chemists, this represents a pivotal shift towards more atom-economical and step-efficient strategies that align with green chemistry principles while maintaining the rigorous purity standards required for pharmaceutical applications.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of quinazolinone derivatives has relied heavily on methodologies that are fraught with operational hazards and economic inefficiencies. Traditional routes often necessitate the use of high-pressure carbon monoxide gas, which requires specialized autoclave reactors and stringent safety protocols, thereby increasing capital expenditure and limiting scalability in standard laboratory or pilot plant settings. Furthermore, many established protocols depend on expensive ruthenium or platinum catalysts, or require the pre-synthesis of complex intermediates such as nitro-substituted benzamides, which adds multiple steps to the overall process and reduces the overall yield. These conventional methods frequently suffer from narrow substrate scopes, failing to tolerate sensitive functional groups like halogens or trifluoromethyl groups, which limits their utility in the late-stage functionalization of drug candidates.
The Novel Approach
In stark contrast, the novel palladium-catalyzed carbonylation cascade described in the patent data offers a robust and versatile alternative that operates under significantly milder conditions. By employing molybdenum hexacarbonyl as a solid carbon monoxide surrogate, the reaction eliminates the safety risks associated with gaseous CO while ensuring a steady release of the carbonyl source in situ. The core transformation involves the direct coupling of trifluoroethylimidoyl chloride with nitro compounds in a one-pot fashion, facilitated by a palladium catalyst system comprising PdCl2 and a dppp ligand. This streamlined approach, illustrated in the reaction scheme below, allows for the direct assembly of the quinazolinone core without the need for isolating unstable intermediates, thereby reducing waste generation and processing time.
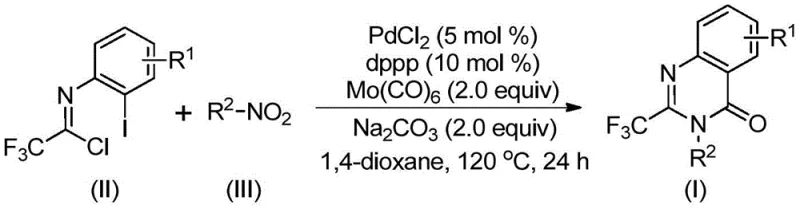
The operational simplicity of this method is further enhanced by the use of common organic solvents like 1,4-dioxane and standard heating conditions at 120°C, making it highly adaptable for both small-scale discovery and larger commercial production. The ability to utilize cheap and commercially available nitro compounds as the nitrogen source is a particular strategic advantage, as it bypasses the need for costly aniline derivatives or pre-formed amides. This shift in synthetic logic not only lowers the raw material costs but also opens up new avenues for structural diversification, enabling chemists to access a wider chemical space for structure-activity relationship studies with greater speed and efficiency.
Mechanistic Insights into Pd-Catalyzed Carbonylation Cascade
The success of this multi-component reaction relies on a sophisticated catalytic cycle that orchestrates several distinct chemical transformations within a single vessel. The mechanism is believed to initiate with the reduction of the nitro group to an amine by molybdenum hexacarbonyl, which simultaneously serves as the reducing agent and the carbon monoxide source. Following this reduction, a base-promoted intermolecular coupling occurs between the newly formed amine and the trifluoroethylimidoyl chloride, generating a trifluoroacetamidine derivative in situ. The palladium catalyst then inserts into the carbon-iodine bond of the imidoyl chloride moiety, forming a key divalent palladium intermediate that sets the stage for ring closure.
Subsequent insertion of carbon monoxide, released thermally from the molybdenum complex, into the carbon-palladium bond generates an acyl-palladium species. This intermediate undergoes intramolecular cyclization facilitated by the base, leading to the formation of a seven-membered cyclic palladium complex. The final step involves reductive elimination, which releases the desired 2-trifluoromethyl substituted quinazolinone product and regenerates the active palladium catalyst for the next cycle. This intricate dance of redox and organometallic steps ensures high selectivity and minimizes the formation of side products, which is crucial for maintaining a clean impurity profile in pharmaceutical intermediates.
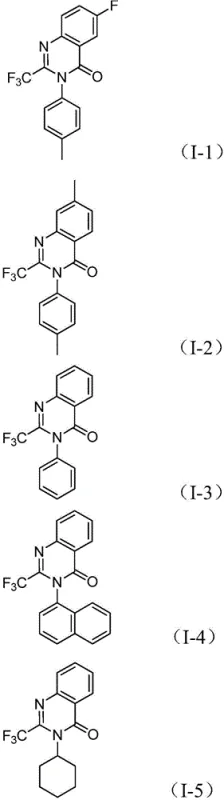
From an impurity control perspective, the one-pot nature of the reaction is highly beneficial as it prevents the accumulation of reactive intermediates that could otherwise degrade or participate in unwanted side reactions. The compatibility of the catalytic system with various electron-withdrawing and electron-donating groups on the aromatic rings, as evidenced by the successful synthesis of derivatives I-1 through I-5, demonstrates the robustness of the mechanism. This broad functional group tolerance ensures that the process can be applied to a wide range of substrates without requiring extensive re-optimization, providing a reliable platform for the synthesis of complex heterocyclic libraries needed for modern drug development programs.
How to Synthesize 2-Trifluoromethyl Quinazolinones Efficiently
The implementation of this synthesis protocol is designed to be straightforward and accessible, requiring standard laboratory equipment and commonly available reagents. The procedure involves charging a reaction vessel with the palladium catalyst, ligand, base, carbon monoxide source, and the two primary organic substrates in an appropriate solvent. The mixture is then heated to the specified temperature for a defined period to ensure complete conversion. Detailed standardized synthesis steps see the guide below.
- Combine palladium chloride, dppp ligand, sodium carbonate, Mo(CO)6, trifluoroethylimidoyl chloride, and nitro compound in an organic solvent like dioxane.
- Heat the reaction mixture to 120°C and stir for 16 to 30 hours to allow the carbonylation cascade to proceed.
- Upon completion, filter the mixture, mix with silica gel, and purify via column chromatography to isolate the target quinazolinone.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain leaders, the adoption of this novel synthetic route offers compelling economic and logistical benefits that directly impact the bottom line. The primary driver for cost reduction lies in the substitution of expensive, pre-activated starting materials with commodity chemicals like nitro compounds and simple imidoyl chlorides. By eliminating the need for high-pressure gas infrastructure and precious metal catalysts like ruthenium or platinum, the process significantly lowers both the capital investment required for equipment and the recurring costs associated with catalyst procurement and disposal. This shift towards cheaper and more abundant raw materials creates a more resilient supply chain that is less susceptible to market volatility and sourcing bottlenecks.
- Cost Reduction in Manufacturing: The economic advantages of this method are derived from the fundamental simplification of the synthetic sequence. By consolidating multiple reaction steps into a single one-pot operation, manufacturers can realize substantial savings in labor, energy consumption, and solvent usage. The avoidance of hazardous high-pressure carbon monoxide gas removes the need for specialized containment systems and safety monitoring, further reducing operational overheads. Additionally, the use of a palladium catalyst system, while still a precious metal, is optimized with ligands that enhance turnover, potentially allowing for lower catalyst loading compared to less efficient traditional methods.
- Enhanced Supply Chain Reliability: The reliance on widely available nitro compounds and commercially sourced trifluoroethylimidoyl chlorides ensures a stable and continuous supply of raw materials. Unlike custom-synthesized intermediates that may have long lead times and limited supplier bases, these starting materials are produced at scale by multiple chemical vendors globally. This diversification of the supply base mitigates the risk of production delays caused by single-source dependencies. Furthermore, the robustness of the reaction conditions means that the process is less sensitive to minor variations in reagent quality, ensuring consistent output even when sourcing from different batches or suppliers.
- Scalability and Environmental Compliance: The transition from batch processes involving hazardous gases to a safer, solution-phase reaction facilitates easier scale-up from gram to kilogram and ton scales. The simplified workup procedure, which involves basic filtration and chromatography, reduces the generation of complex waste streams that require expensive treatment. By minimizing the use of toxic reagents and avoiding high-pressure operations, the process aligns better with increasingly stringent environmental regulations and corporate sustainability goals. This compliance advantage not only reduces regulatory risk but also enhances the marketability of the final product to environmentally conscious pharmaceutical partners.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These answers are derived directly from the experimental data and technical specifications outlined in the patent documentation to provide clarity on process capabilities and limitations. Understanding these details is essential for evaluating the feasibility of integrating this method into existing manufacturing workflows.
Q: Does this synthesis require high-pressure carbon monoxide gas?
A: No, the method utilizes molybdenum hexacarbonyl (Mo(CO)6) as a solid carbon monoxide substitute, eliminating the need for dangerous high-pressure CO gas cylinders and specialized autoclave equipment.
Q: What is the substrate scope for the nitro compound component?
A: The process demonstrates excellent compatibility with various substituents including halogens (F, Cl, Br), alkyl groups (methyl), and trifluoromethyl groups at ortho, meta, or para positions on the aromatic ring.
Q: How is the final product purified after the reaction?
A: The post-treatment involves a straightforward filtration followed by silica gel mixing and standard column chromatography, avoiding complex extraction or crystallization steps often required in traditional methods.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Quinazolinone Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that efficient synthetic methodologies play in accelerating drug development and ensuring commercial viability. Our team of expert process chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory discovery to industrial manufacturing is seamless and robust. We are committed to delivering high-purity pharmaceutical intermediates that meet stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation to verify every batch.
We invite you to collaborate with us to leverage this advanced carbonylation technology for your specific project needs. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your volume requirements and quality targets. Please contact us today to request specific COA data and route feasibility assessments, and let us help you optimize your supply chain for the next generation of quinazolinone-based therapeutics.