Scalable Synthesis of Potent Benzacridine Derivatives Targeting c-myc Oncogene Expression
Scalable Synthesis of Potent Benzacridine Derivatives Targeting c-myc Oncogene Expression
The pharmaceutical industry continuously seeks novel scaffolds capable of disrupting oncogenic pathways with high specificity and low toxicity. Patent CN102351870B discloses a robust methodology for preparing a series of benzacridine derivatives that exhibit potent inhibitory activity against the c-myc proto-oncogene DNA. These compounds are characterized by a fused tetracyclic core where substituents R1 and R2 can be methoxy or methylenedioxy groups, and the side chain at the -position allows for significant structural diversity through amine substitution. This technological breakthrough addresses the critical need for effective anticancer agents that can selectively target DNA structures rich in guanine, offering a promising avenue for drug development. The structural versatility of these molecules allows for fine-tuning of pharmacokinetic properties, making them highly attractive candidates for further preclinical evaluation.
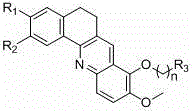
As a reliable pharmaceutical intermediate supplier, understanding the synthetic accessibility of such complex heterocycles is paramount for ensuring supply chain continuity. The disclosed method provides a clear pathway from commodity chemicals to high-value active pharmaceutical ingredients (APIs). By leveraging established organic transformations such as Friedel-Crafts acylation and cyclodehydration, the process minimizes the reliance on exotic catalysts while maintaining high regioselectivity. This balance between chemical complexity and operational simplicity is essential for manufacturers aiming to reduce lead time for high-purity pharmaceutical intermediates. The ability to introduce diverse amine functionalities in the final steps further enhances the commercial viability of this platform, allowing for the rapid generation of analog libraries for structure-activity relationship (SAR) studies.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional syntheses of acridine-based anticancer agents often suffer from harsh reaction conditions that compromise yield and purity. Conventional routes may require multiple protection and deprotection steps to manage the reactivity of phenolic hydroxyl groups, leading to increased waste generation and higher production costs. Furthermore, achieving the correct regiochemistry during the ring-closing steps can be challenging, often resulting in difficult-to-separate isomeric impurities that complicate downstream purification. Many existing methods rely on expensive transition metal catalysts or unstable intermediates that pose safety risks during commercial scale-up of complex pharmaceutical intermediates. These inefficiencies create bottlenecks in the supply chain, making it difficult to secure consistent quantities of high-quality material for clinical trials.
The Novel Approach
The methodology outlined in the patent introduces a streamlined strategy that overcomes these historical hurdles through a logical sequence of bond-forming reactions. By initiating the synthesis with the acylation of 1,2-dimethoxybenzene, the process establishes the carbon skeleton efficiently before introducing nitrogen functionality. The use of polyphosphoric acid for cyclization offers a powerful dehydrating environment that drives the formation of the tetralone core with excellent conversion rates. Subsequent functionalization steps, including demethylation and coupling with protected amino-benzoic acids, are optimized to preserve the integrity of the sensitive acridine nucleus. This approach not only simplifies the overall workflow but also enhances the robustness of the process, making it suitable for cost reduction in API manufacturing.
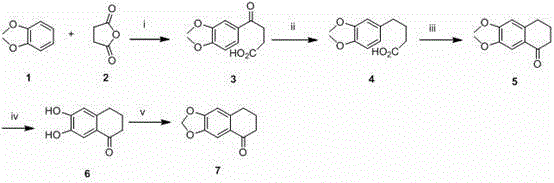
Mechanistic Insights into POCl3-Mediated Cyclization and Coupling
The core construction of the benzacridine skeleton relies heavily on the efficient coupling of the tetralone fragment with the aniline derivative. In the presence of phosphorus oxychloride (POCl3), the carboxylic acid group of the aniline precursor is activated to form a reactive acyl chloride or mixed anhydride species in situ. This activated intermediate then undergoes nucleophilic attack by the enolizable ketone of the tetralone moiety, followed by dehydration and aromatization to close the central pyridine ring. The reaction temperature is carefully controlled, typically refluxing for several hours to ensure complete conversion while minimizing thermal degradation of the product. This mechanistic pathway is favored because it avoids the use of external coupling reagents that could introduce difficult-to-remove impurities into the final drug substance.
Impurity control is a critical aspect of this synthesis, particularly regarding the selectivity of the alkylation steps. When introducing the side chain via dibromoalkanes, the reaction conditions utilizing potassium carbonate in acetonitrile promote SN2 substitution at the primary bromide while leaving the secondary positions untouched. The subsequent displacement of the terminal bromide by various amines (such as pyrrolidine, piperidine, or morpholine) is highly chemoselective, ensuring that the nitrogen atom attaches exclusively to the intended alkyl chain. This precision is vital for maintaining the biological activity of the final compound, as even minor structural deviations can significantly alter DNA binding affinity. The rigorous purification protocols described, including ion exchange resin treatment to swap triflate for chloride counterions, further guarantee the high purity required for pharmaceutical applications.
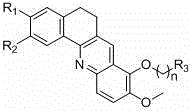
How to Synthesize Benzacridine Derivatives Efficiently
The synthesis of these potent anticancer agents follows a convergent strategy that maximizes yield at each stage. The process begins with the preparation of the tetralone building block, followed by the separate synthesis of the functionalized aniline coupling partner. These two fragments are then united under dehydrating conditions to form the tricyclic intermediate, which is subsequently elaborated to the final tetracyclic product. Detailed operational parameters, such as solvent ratios and temperature profiles, are critical for reproducibility. For a comprehensive guide on executing these transformations in a GMP environment, please refer to the standardized protocol below.
- Perform Friedel-Crafts acylation of 1,2-dimethoxybenzene with succinic anhydride using AlCl3 catalyst to form the tetralone scaffold.
- Execute deoxygenation using triethylsilane and trifluoroacetic acid, followed by cyclization with polyphosphoric acid to close the ring system.
- Conduct demethylation with HBr, protect phenolic groups, and couple with amino-benzoic acid derivatives using POCl3 to form the acridine core.
- Finalize the synthesis by substituting the bromo-alkoxy side chain with various amines to generate the target benzacridine derivatives.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement perspective, this synthetic route offers significant advantages due to its reliance on widely available starting materials. Commodities like veratrole, succinic anhydride, and o-vanillin are produced on a massive industrial scale, ensuring a stable supply base that is less susceptible to market volatility. The elimination of rare earth catalysts or proprietary ligands in the key bond-forming steps reduces the dependency on single-source suppliers, thereby enhancing supply chain reliability. Moreover, the use of common solvents such as nitrobenzene, dichloromethane, and acetonitrile simplifies waste management and solvent recovery processes, contributing to a more sustainable manufacturing footprint. These factors collectively lower the barrier to entry for commercial production.
- Cost Reduction in Manufacturing: The process achieves cost efficiency by minimizing the number of isolation steps and utilizing high-yielding transformations. For instance, the reduction of the ketone intermediate using triethylsilane and trifluoroacetic acid proceeds with nearly quantitative conversion, reducing material loss. Additionally, the avoidance of chromatographic purification in favor of recrystallization for key intermediates significantly lowers processing costs. By streamlining the synthesis to fewer operational units, manufacturers can achieve substantial cost savings without compromising on the quality of the final active ingredient.
- Enhanced Supply Chain Reliability: The modular nature of the synthesis allows for the parallel production of different intermediates, which mitigates the risk of bottlenecks. If a specific amine side chain is required, it can be introduced in the final step without altering the upstream production of the core scaffold. This flexibility ensures that production schedules can be adjusted rapidly to meet changing demand from clinical partners. Furthermore, the robustness of the reaction conditions means that the process can be transferred between different manufacturing sites with minimal re-validation, securing long-term supply continuity.
- Scalability and Environmental Compliance: The reactions described are inherently scalable, having been demonstrated effectively from gram to multi-gram scales in the patent examples. The use of aqueous workups and standard extraction techniques facilitates easy scale-up to tonnage production. Environmental compliance is supported by the ability to recover and recycle solvents like DMF and acetonitrile, and the absence of heavy metal catalysts in the final steps simplifies the removal of toxic residues. This alignment with green chemistry principles makes the process attractive for modern pharmaceutical manufacturing facilities focused on sustainability.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these benzacridine derivatives. The answers are derived directly from the experimental data and technical specifications provided in the patent documentation. Understanding these details is crucial for R&D teams evaluating the feasibility of incorporating this scaffold into their drug discovery pipelines.
Q: What is the primary biological target of these benzacridine derivatives?
A: These derivatives are designed to interact strongly with c-myc proto-oncogene DNA, specifically inhibiting its expression and demonstrating significant antitumor effects against cell lines like JEG-3 and HeLa.
Q: How does this synthesis method improve upon conventional routes?
A: The patented method utilizes a modular approach starting from readily available veratrole and succinic anhydride, avoiding complex protecting group strategies in early stages and enabling high-yield cyclization via polyphosphoric acid.
Q: Is this process suitable for large-scale commercial production?
A: Yes, the reaction conditions utilize standard industrial reagents like aluminum chloride and phosphorus oxychloride, and the purification steps rely on common techniques like recrystallization and column chromatography, facilitating scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Benzacridine Derivative Supplier
The development of novel anticancer agents requires a partner who understands both the scientific intricacies and the commercial realities of pharmaceutical manufacturing. NINGBO INNO PHARMCHEM possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project can transition smoothly from the lab to the market. Our stringent purity specifications and rigorous QC labs guarantee that every batch of benzacridine derivative meets the highest international standards, providing you with the confidence needed to advance your clinical programs. We are committed to supporting your innovation with reliable supply and technical expertise.
We invite you to discuss how our capabilities can support your specific requirements for high-purity pharmaceutical intermediates. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your volume needs. Please contact us to request specific COA data and route feasibility assessments, and let us help you accelerate the development of your next-generation cancer therapy.