Scalable Synthesis of Novel Benzacridine Derivatives for Oncology Drug Development
Scalable Synthesis of Novel Benzacridine Derivatives for Oncology Drug Development
The pharmaceutical industry is constantly seeking novel scaffolds that can effectively target oncogenic drivers with high specificity and low toxicity. Patent CN102351870A introduces a significant advancement in this domain by disclosing a series of benzacridine derivatives designed to inhibit the c-myc proto-oncogene. This genetic target is crucial in cell proliferation and is frequently dysregulated in various human cancers. The disclosed compounds feature a unique tetracyclic core structure where substituents R1 and R2 can be methoxy or methylenedioxy groups, and the side chain at position R3 allows for extensive diversification with amines. This structural flexibility is paramount for optimizing pharmacokinetic properties and binding affinity. For R&D directors and procurement specialists, understanding the synthetic accessibility of such complex heterocycles is critical for supply chain planning. The patent outlines a robust, multi-step pathway that transforms simple commodity chemicals into high-value anticancer intermediates, demonstrating a clear path from bench-scale discovery to potential commercial production.
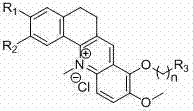
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthesis of acridine and benzacridine skeletons often relies on harsh conditions that pose significant challenges for industrial scale-up. Classical methods frequently involve the Bernthsen acridine synthesis, which requires heating diphenylamine derivatives with carboxylic acids at very high temperatures, often exceeding 200°C, leading to poor atom economy and difficult purification profiles. Furthermore, many existing routes utilize expensive or hazardous reagents that complicate waste management and increase the overall cost of goods sold (COGS). The formation of the central nitrogen-containing ring system can also suffer from low regioselectivity, generating isomeric impurities that are difficult to separate without extensive chromatography. These factors collectively result in long lead times and unstable supply chains for critical oncology intermediates. Additionally, the introduction of specific functional groups required for DNA intercalation often requires late-stage modifications that are low-yielding, further exacerbating cost inefficiencies in the manufacturing process.
The Novel Approach
The methodology presented in CN102351870A offers a strategic departure from these legacy issues by employing a convergent synthesis strategy centered on Friedel-Crafts chemistry and modern cyclization techniques. The route begins with the acylation of 1,2-dimethoxybenzene (veratrole) with succinic anhydride, a reaction that is well-understood and easily controlled. By utilizing aluminum chloride as a Lewis acid catalyst at moderate temperatures ranging from 0°C to 60°C, the process ensures high selectivity for the desired ketone intermediate. Subsequent reduction and intramolecular cyclization steps build the tetralone core efficiently. The key innovation lies in the condensation of this core with a protected vanillin derivative using phosphorus oxychloride (POCl3) to form the pyridine ring, followed by catalytic hydrogenation to remove chlorine atoms. This sequence not only improves overall yields but also allows for the precise installation of the side chains necessary for biological activity. The modularity of this approach means that different amine functionalities can be introduced in the final steps, facilitating rapid structure-activity relationship (SAR) studies without redesigning the entire synthetic route.
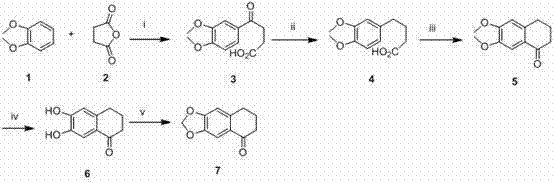
Mechanistic Insights into POCl3-Mediated Cyclization and Functionalization
The core construction of the benzacridine scaffold relies heavily on the efficiency of the ring-closing step mediated by phosphorus oxychloride. In this mechanism, the carbonyl oxygen of the tetralone intermediate and the amino group of the aniline derivative undergo a condensation reaction. POCl3 acts as a dehydrating agent and activator, converting the hydroxyl groups into good leaving groups, thereby facilitating the nucleophilic attack that closes the pyridine ring. This step typically proceeds under reflux conditions for approximately 8 hours, ensuring complete conversion. The resulting chloro-intermediate is then subjected to catalytic hydrogenation using palladium on carbon (Pd/C) in a mixed solvent system of DMF and ethanol. This hydrodechlorination is critical as it removes the chlorine atom introduced during cyclization, restoring the aromaticity and preparing the molecule for subsequent functionalization. The use of 5-10% Pd/C at 60°C under a hydrogen atmosphere provides a clean transformation with minimal over-reduction of other sensitive functional groups. Following this, the phenolic hydroxyl group, generated by hydrolysis of the sulfonate protecting group using sodium hydroxide at 100°C, serves as the anchor point for the alkyl side chain. This sequential logic ensures that each functional group is manipulated at the optimal stage of the synthesis, minimizing side reactions and maximizing purity.
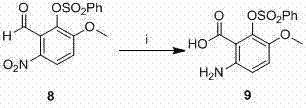
Impurity control is inherently built into this synthetic design through the use of crystallization and selective precipitation techniques described in the embodiments. For instance, the initial acylation product is purified by recrystallization from ethyl acetate, which effectively removes unreacted starting materials and poly-acylated byproducts. Similarly, the intermediate ketones and final benzacridine salts are isolated via filtration after pouring reaction mixtures into ice water, a technique that leverages solubility differences to achieve high purity without the need for preparative HPLC. The patent specifies rigorous molar ratios, such as maintaining a 1:1.5 to 1:2.5 ratio between the tetralone and aniline components, to prevent the accumulation of unreacted precursors. Furthermore, the final ion exchange step, where triflate anions are exchanged for chloride ions using anion exchange resin, ensures that the final drug substance meets stringent salt form specifications required for pharmaceutical formulations. This attention to downstream processing details highlights the process's readiness for GMP manufacturing environments.
How to Synthesize Benzacridine Derivatives Efficiently
The synthesis of these potent anticancer agents involves a carefully orchestrated sequence of organic transformations that balance reactivity with selectivity. The process initiates with the formation of the bicyclic ketone core, followed by its condensation with a functionalized aniline to construct the tricyclic acridine system. Detailed operational parameters, including specific solvent volumes, temperature ramps, and workup procedures, are essential for reproducing the high yields reported in the patent embodiments. For process chemists looking to implement this route, understanding the nuances of the reduction and alkylation steps is vital for success. The following guide outlines the standardized synthesis steps derived directly from the patent data to ensure reproducibility and safety in the laboratory.
- Perform Friedel-Crafts acylation of 1,2-dimethoxybenzene with succinic anhydride using AlCl3 catalyst at 0-60°C.
- Execute reduction of the keto-acid intermediate using triethylsilane/trifluoroacetic acid, followed by cyclization with polyphosphoric acid.
- Conduct condensation with protected vanillin derivatives using POCl3, followed by catalytic hydrogenation and alkylation to finalize the structure.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement perspective, the synthetic route detailed in CN102351870A presents several compelling advantages that translate directly into cost reduction in pharmaceutical intermediate manufacturing. The reliance on bulk commodity chemicals such as succinic anhydride, veratrole, and vanillin derivatives ensures a stable and predictable supply chain, mitigating the risks associated with sourcing exotic or single-source reagents. These starting materials are produced globally at massive scales, which inherently drives down their unit cost and guarantees availability even during market fluctuations. Moreover, the reaction conditions employed, such as the use of nitrobenzene and DMF, utilize solvents that are standard in the fine chemical industry, simplifying solvent recovery and recycling protocols. This compatibility with existing infrastructure means that contract development and manufacturing organizations (CDMOs) can adopt this process with minimal capital expenditure on new equipment. The elimination of cryogenic conditions and the use of moderate temperatures throughout the synthesis further reduce energy consumption, contributing to a lower carbon footprint and reduced utility costs.
- Cost Reduction in Manufacturing: The process achieves significant cost optimization by avoiding the use of precious metal catalysts in the early stages, reserving palladium only for the specific dechlorination step where it is strictly necessary. Since palladium is a high-cost input, limiting its usage to a single catalytic step rather than multiple stoichiometric steps drastically reduces the raw material bill. Additionally, the high yields reported in the embodiments, such as the 97.7% yield in the reduction step and 93.3% in the cyclization step, minimize material loss and waste disposal costs. The ability to purify intermediates via simple filtration and recrystallization rather than column chromatography on a large scale further enhances economic efficiency. These factors combined create a highly competitive cost structure for the final active pharmaceutical ingredient (API).
- Enhanced Supply Chain Reliability: The modular nature of the synthesis allows for the decoupling of production stages, enabling manufacturers to stockpile key intermediates like the tetralone core or the protected aniline derivative. This strategy buffers against potential disruptions in the supply of specific reagents, ensuring continuous production flow. The use of robust chemical transformations that are tolerant to minor variations in reaction parameters adds another layer of reliability, reducing the likelihood of batch failures. Furthermore, the final alkylation step allows for the late-stage introduction of diverse amine side chains, meaning a single batch of the core benzacridine scaffold can be diverted to produce multiple different drug candidates. This flexibility is invaluable for supply chain managers who need to respond quickly to changing clinical demands without requalifying entirely new synthetic routes.
- Scalability and Environmental Compliance: The synthetic pathway is designed with scalability in mind, utilizing reaction vessels and conditions that are easily transferable from pilot plant to commercial production scales. The avoidance of highly toxic reagents like phosgene or azides simplifies environmental health and safety (EHS) compliance, reducing the regulatory burden on manufacturing sites. Waste streams generated during the process, primarily consisting of aqueous acidic or basic washes, can be treated using standard neutralization and extraction protocols familiar to wastewater treatment facilities. The high atom economy of the condensation and cyclization steps ensures that the majority of the mass of the starting materials ends up in the final product, aligning with green chemistry principles. This environmental stewardship not only protects the ecosystem but also safeguards the manufacturer against future regulatory tightening regarding chemical emissions.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these benzacridine derivatives. These answers are synthesized from the detailed experimental data and background information provided in the patent documentation. They serve to clarify the feasibility of the technology for potential partners and investors. Understanding these aspects is crucial for making informed decisions about licensing or contracting the manufacturing of these compounds.
Q: What is the primary biological target of these benzacridine derivatives?
A: The derivatives are designed to strongly inhibit the expression of the c-myc proto-oncogene DNA, showing significant antitumor effects against cell lines like JEG-3 and HeLa.
Q: How does this synthesis route improve upon conventional acridine production?
A: This method utilizes accessible starting materials like succinic anhydride and veratrole, employing robust Friedel-Crafts chemistry that avoids extremely harsh conditions often found in traditional acridine synthesis.
Q: Is the process suitable for large-scale commercial manufacturing?
A: Yes, the reaction conditions (0-60°C for acylation, standard reflux for cyclization) and the use of common solvents like nitrobenzene and DMF indicate high feasibility for scale-up from kilogram to tonnage levels.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Benzacridine Derivative Supplier
The technological potential of the benzacridine derivatives described in CN102351870A is immense, offering a promising avenue for the development of next-generation anticancer therapeutics. However, translating this patented chemistry into a commercial reality requires a partner with deep expertise in process optimization and scale-up. NINGBO INNO PHARMCHEM stands ready to support your drug development pipeline with our extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our state-of-the-art facilities are equipped to handle the specific solvent systems and reaction conditions required for this synthesis, ensuring that stringent purity specifications are met consistently. With our rigorous QC labs and commitment to quality, we guarantee that every batch of high-purity pharmaceutical intermediates delivered meets the highest international standards, providing you with the confidence needed to advance your clinical trials.
We invite you to collaborate with us to unlock the full commercial value of this innovative technology. Our technical team is prepared to conduct a Customized Cost-Saving Analysis tailored to your specific volume requirements, identifying further opportunities for efficiency gains. We encourage you to contact our technical procurement team today to request specific COA data and route feasibility assessments. By partnering with us, you gain access to a reliable supply chain that prioritizes both quality and speed, ensuring that your life-saving medications reach patients without delay. Let us be your trusted ally in navigating the complexities of fine chemical manufacturing and bringing this breakthrough oncology treatment to the global market.