Advanced Metal-Free Synthesis of Gem-Difluoroolefins for Commercial Scale-Up
The pharmaceutical and agrochemical industries are constantly seeking efficient pathways to incorporate fluorine into organic scaffolds, driven by the unique ability of fluorine atoms to modulate metabolic stability, lipophilicity, and bioavailability. A significant breakthrough in this domain is documented in patent CN114409515A, which discloses a novel preparation method for gem-difluoroolefin compounds. This technology represents a paradigm shift from classical carbonyl olefination strategies to a more streamlined hydrodefluorination approach. By utilizing trifluoromethyl olefin compounds as precursors, the method achieves the selective removal of a single fluorine atom to generate the valuable gem-difluoroalkenyl motif. For R&D directors and procurement specialists, this innovation offers a compelling alternative to traditional synthesis routes, promising enhanced purity profiles and simplified supply chains for high-purity pharmaceutical intermediates. The strategic implementation of this chemistry allows manufacturers to access complex fluorinated building blocks with greater reliability and reduced environmental impact.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of gem-difluoroolefin structures has relied heavily on classical carbon-carbon bond-forming reactions such as the Wittig reaction or the Horner-Wadsworth-Emmons (HWE) reaction. While these methods are foundational in organic synthesis, they present significant drawbacks when applied to complex, multifunctional molecules typical in modern drug discovery. These conventional protocols typically necessitate the use of strong bases to generate reactive ylides or carbanions, conditions that are often incompatible with base-sensitive functional groups such as esters, nitriles, or certain heterocycles. Furthermore, the stereoselectivity of these reactions can be difficult to control, often resulting in mixtures of E/Z isomers that require tedious and yield-losing separation processes. The reliance on specialized phosphorus reagents and the generation of stoichiometric phosphine oxide waste also pose challenges for cost reduction in pharmaceutical intermediate manufacturing, particularly when scaling to multi-kilogram quantities where waste disposal becomes a major logistical and financial burden.
The Novel Approach
In stark contrast to the harsh conditions of the past, the methodology described in CN114409515A introduces a mild, metal-free hydrodefluorination strategy that elegantly bypasses these limitations. The core of this innovation lies in the use of diphenylphosphine oxide as a reducing agent in conjunction with cesium carbonate as a base, facilitated by a specific amount of water as an additive. This system operates effectively in ethyl acetate at a moderate temperature of 70°C, providing a reaction environment that is remarkably tolerant of diverse functional groups. As illustrated in the reaction scheme below, the transformation converts readily available trifluoromethyl olefins into the desired gem-difluoroolefins with high regioselectivity. This approach not only simplifies the synthetic route by avoiding the need for pre-functionalized carbonyl precursors but also eliminates the requirement for expensive transition metal catalysts, thereby streamlining the downstream purification process and enhancing the overall economic viability of the synthesis.
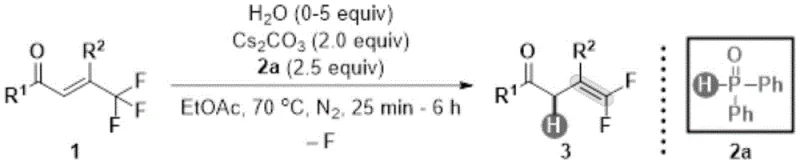
Mechanistic Insights into Diphenylphosphine Oxide Promoted Hydrodefluorination
The mechanistic elegance of this transformation is rooted in the unique reactivity of the diphenylphosphine oxide and cesium carbonate system. Unlike transition-metal catalyzed defluorination which often proceeds through oxidative addition and reductive elimination cycles involving expensive palladium or nickel complexes, this metal-free protocol likely operates through a nucleophilic activation pathway. The cesium carbonate serves to deprotonate the diphenylphosphine oxide, generating a nucleophilic phosphinate species that can interact with the electron-deficient trifluoromethyl olefin. The presence of water is critical, acting as a proton source to facilitate the final elimination of the fluoride ion and regeneration of the active phosphine oxide species or formation of the phosphate byproduct. This mechanism ensures that the reaction proceeds with excellent chemoselectivity, leaving other sensitive groups on the aromatic rings—such as bromides, chlorides, or nitro groups—intact. For the R&D director, understanding this mechanism is vital for impurity control; the absence of metal residues means that the stringent purity specifications required for API intermediates can be met with standard crystallization or chromatography techniques, avoiding the need for specialized metal scavenging resins.
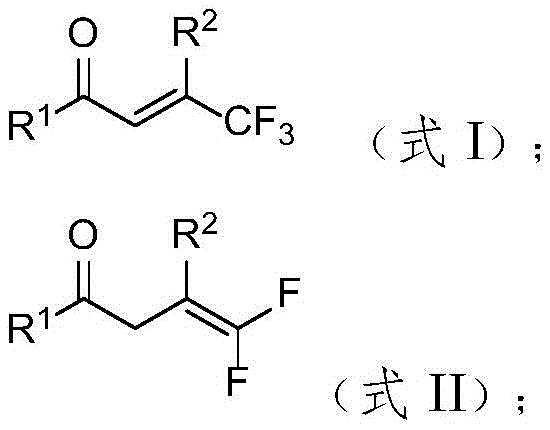
How to Synthesize Gem-Difluoroolefin Efficiently
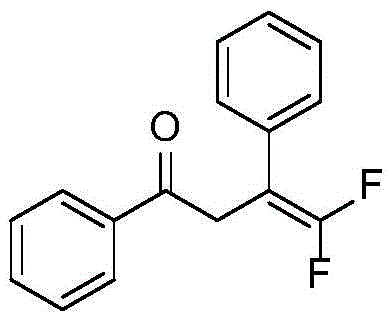
Implementing this synthesis in a laboratory or pilot plant setting requires strict adherence to the optimized parameters identified in the patent data to ensure maximum yield and reproducibility. The process begins with the precise weighing of the trifluoromethyl olefin substrate, diphenylphosphine oxide, and cesium carbonate, followed by the addition of ethyl acetate as the solvent. A critical step is the controlled addition of water, which acts as a reaction promoter; optimization studies indicate that 5 equivalents of water provide the optimal balance for reaction kinetics, whereas excessive water can lead to diminished returns. The reaction is conducted under an inert nitrogen atmosphere to prevent oxidation of the phosphine reagent and is heated to 70°C for a duration ranging from 25 minutes to 6 hours, depending on the specific electronic nature of the substrate. Upon completion, the workup involves a standard aqueous quench and extraction protocol, followed by purification via silica gel column chromatography to isolate the high-purity product. The specific structure of a representative product, 4,4-difluoro-1,3-diphenyl-3-buten-1-one, is shown below, exemplifying the clean conversion achievable with this method.
- Combine trifluoromethyl olefin substrate, diphenylphosphine oxide (2.5 equiv), cesium carbonate (2.0 equiv), and water (5.0 equiv) in ethyl acetate solvent under nitrogen atmosphere.
- Heat the reaction mixture to 70°C and stir for 25 minutes to 6 hours to facilitate the hydrodefluorination process.
- Quench with saturated ammonium chloride, extract with ethyl acetate, dry over sodium sulfate, and purify via silica gel column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this hydrodefluorination technology offers tangible benefits that extend beyond mere chemical curiosity. The primary advantage lies in the drastic simplification of the raw material portfolio; by utilizing trifluoromethyl olefins as direct precursors, manufacturers can bypass multi-step sequences traditionally required to install the difluoroalkene moiety. This consolidation of steps translates directly into substantial cost savings and reduced lead time for high-purity pharmaceutical intermediates. Furthermore, the elimination of transition metal catalysts removes a significant bottleneck in the supply chain, as there is no longer a dependency on volatile precious metal markets or the need for complex metal removal validation studies. The use of ethyl acetate, a common and environmentally benign solvent, further aligns with green chemistry initiatives, potentially reducing waste disposal costs and easing regulatory compliance burdens associated with volatile organic compound (VOC) emissions. This robust and scalable process ensures a reliable gem-difluoroolefin supplier can maintain continuous production schedules even amidst fluctuating market demands.
- Cost Reduction in Manufacturing: The economic impact of this method is profound, primarily driven by the replacement of expensive transition metal catalysts with inexpensive, commodity-grade reagents like cesium carbonate and diphenylphosphine oxide. By removing the need for palladium or nickel, the process eliminates the costly downstream purification steps associated with heavy metal clearance, such as the use of specialized scavenger resins or repeated recrystallizations. Additionally, the high atom economy of the hydrodefluorination reaction minimizes waste generation, leading to lower disposal fees and a more efficient use of raw materials. The mild reaction conditions also reduce energy consumption compared to high-temperature or cryogenic alternatives, contributing to a leaner and more cost-effective manufacturing profile that enhances overall profit margins.
- Enhanced Supply Chain Reliability: From a logistics perspective, the reliance on widely available and stable reagents significantly de-risks the supply chain. Diphenylphosphine oxide and cesium carbonate are bulk chemicals with established global supply networks, ensuring that production is not held hostage by the scarcity of specialized ligands or catalysts. The robustness of the reaction against moisture and air (to a reasonable extent under nitrogen) simplifies operational requirements, allowing for flexible scheduling and reduced downtime. This stability ensures that commercial scale-up of complex pharmaceutical intermediates can proceed without the frequent interruptions often caused by reagent degradation or supply shortages, guaranteeing consistent delivery timelines to downstream partners.
- Scalability and Environmental Compliance: The scalability of this process is supported by its simplicity and the use of standard unit operations familiar to any chemical manufacturing facility. The reaction does not require exotic high-pressure equipment or cryogenic cooling, making it easily adaptable from gram-scale laboratory synthesis to ton-scale commercial production. Environmentally, the choice of ethyl acetate as a solvent is a strategic advantage; it is biodegradable and has a favorable safety profile compared to chlorinated solvents often used in fluorine chemistry. This alignment with green chemistry principles facilitates easier permitting and regulatory approval, positioning the manufacturer as a responsible partner committed to sustainable practices while maintaining high throughput capabilities.
Frequently Asked Questions (FAQ)
To assist technical teams in evaluating the feasibility of this technology for their specific projects, we have compiled answers to common inquiries regarding the reaction scope and operational parameters. These insights are derived directly from the experimental data provided in the patent literature, ensuring that the information is grounded in verified scientific results. Understanding these nuances is critical for project planning and risk assessment, particularly when integrating new synthetic routes into existing manufacturing workflows. The following questions address the key differentiators of this method compared to legacy technologies.
Q: What are the advantages of this hydrodefluorination method over traditional Wittig reactions?
A: Unlike traditional Wittig or Horner-Wadsworth-Emmons reactions that require strong basic conditions and often suffer from limited substrate tolerance, this novel method operates under mild conditions (70°C) with excellent functional group compatibility, allowing for the preservation of sensitive moieties like esters and halogens.
Q: Is this synthesis compatible with large-scale manufacturing?
A: Yes, the process utilizes commercially available reagents such as cesium carbonate and diphenylphosphine oxide in ethyl acetate, a green solvent. The absence of expensive transition metal catalysts simplifies the purification process, making it highly suitable for cost reduction in pharmaceutical intermediate manufacturing and scalable production.
Q: What is the role of water in this reaction system?
A: Water acts as a crucial reaction promoter or additive. Optimization data indicates that adding approximately 5 equivalents of water significantly enhances the reaction yield compared to anhydrous conditions, likely by facilitating the protonation steps necessary for the hydrodefluorination mechanism without degrading the reagents.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Gem-Difluoroolefin Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of the hydrodefluorination technology described in CN114409515A for the next generation of fluorinated therapeutics. As a premier CDMO partner, we possess the technical expertise and infrastructure to translate this innovative academic research into robust, commercial-grade manufacturing processes. Our team has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from benchtop discovery to market supply is seamless and efficient. We are equipped with rigorous QC labs and advanced analytical instrumentation to meet stringent purity specifications, guaranteeing that every batch of gem-difluoroolefin intermediate delivered meets the highest industry standards for quality and consistency.
We invite you to collaborate with us to leverage this cutting-edge synthesis for your pipeline. By partnering with our technical procurement team, you can obtain a Customized Cost-Saving Analysis tailored to your specific molecule, demonstrating exactly how this metal-free route can optimize your budget. We encourage you to reach out today to request specific COA data and route feasibility assessments, allowing us to demonstrate our commitment to being your trusted partner in advancing complex fluorine chemistry from concept to commercial reality.