Advanced Synthesis of 5-Trifluoromethyl-1,2,4-Triazoles for Scalable Pharmaceutical Manufacturing
Advanced Synthesis of 5-Trifluoromethyl-1,2,4-Triazoles for Scalable Pharmaceutical Manufacturing
The integration of fluorine atoms, particularly the trifluoromethyl group, into heterocyclic scaffolds has become a cornerstone strategy in modern medicinal chemistry and agrochemical design. As detailed in the groundbreaking patent CN110467579B, a novel and highly efficient preparation method for 5-trifluoromethyl substituted 1,2,4-triazole compounds has been developed, addressing critical bottlenecks in the supply chain of high-value intermediates. The 1,2,4-triazole motif is ubiquitous in bioactive molecules, serving as a key pharmacophore in numerous therapeutic agents ranging from antifungal medications to kinase inhibitors, as illustrated by the diverse biological applications shown in the reference structures below.
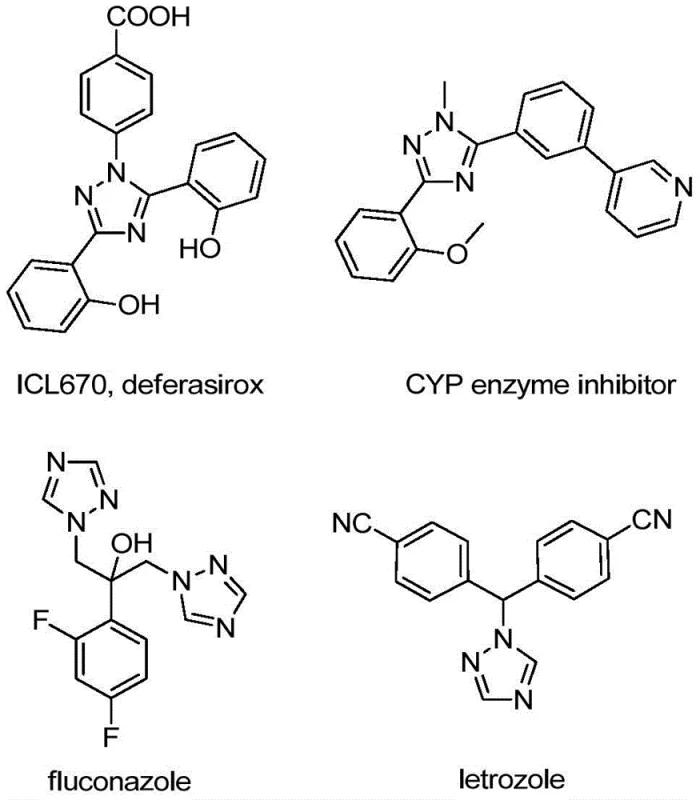
This patented technology offers a transformative approach to constructing these valuable nitrogen-containing five-membered heterocycles. By leveraging a non-metallic iodine-promoted cyclization strategy, the process circumvents the limitations of traditional methods that often rely on hazardous reagents or complex catalytic systems. For R&D directors and process chemists, this represents a significant opportunity to streamline the synthesis of complex APIs, ensuring higher purity profiles and reduced impurity burdens. The ability to introduce the trifluoromethyl group directly during the ring-forming step, rather than through post-synthetic modification, enhances atom economy and simplifies the overall synthetic route, making it an attractive candidate for cost reduction in pharmaceutical intermediate manufacturing.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of trifluoromethyl-substituted nitrogen heterocycles has been fraught with significant technical and economic challenges that hinder large-scale production. Conventional strategies typically fall into two categories: the direct trifluoromethylation of pre-synthesized heterocycles or the cycloaddition of trifluoromethyl-containing synthons. The former often necessitates the use of expensive and specialized trifluoromethylating reagents, such as Togni or Umemoto reagents, which drastically inflate raw material costs and generate substantial stoichiometric waste. Furthermore, alternative methods utilizing trifluorodiazoethane pose severe safety risks due to the explosive nature of diazo compounds, requiring specialized equipment and rigorous safety protocols that are difficult to maintain in a multi-ton commercial setting.
Additionally, many existing protocols rely heavily on transition metal catalysts, such as copper or palladium complexes, to facilitate the coupling reactions. While effective on a milligram scale, these metal-catalyzed processes introduce significant complications during scale-up, particularly regarding the removal of trace metal residues to meet stringent regulatory limits for active pharmaceutical ingredients (APIs). The requirement for strict anhydrous and anaerobic conditions in many of these traditional methods further exacerbates operational costs, demanding inert gas lines, dried solvents, and glovebox techniques that are impractical for high-throughput manufacturing. These cumulative factors result in prolonged lead times and reduced overall process efficiency, creating a bottleneck for the reliable supply of high-purity pharmaceutical intermediates.
The Novel Approach
In stark contrast to these cumbersome legacy methods, the invention disclosed in patent CN110467579B introduces a streamlined, iodine-promoted synthesis that fundamentally reshapes the production landscape for 5-trifluoromethyl-1,2,4-triazoles. This innovative route utilizes readily available and inexpensive starting materials, specifically trifluoroethylimidoyl chloride and hydrazones, which are combined in the presence of sodium acetate and elemental iodine. The reaction proceeds smoothly in common organic solvents like 1,2-dichloroethane (DCE) at moderate temperatures, typically around 80°C, eliminating the need for cryogenic conditions or high-pressure reactors. The general reaction scheme depicted below highlights the simplicity and elegance of this transformation, where the trifluoromethyl group is incorporated directly from the imidoyl chloride synthon.
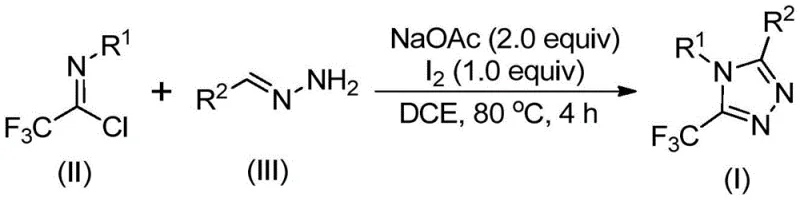
The operational simplicity of this novel approach cannot be overstated, as it functions effectively under ambient atmospheric conditions without the necessity for rigorous exclusion of moisture or oxygen. This robustness significantly lowers the barrier to entry for commercial scale-up, allowing manufacturers to utilize standard reactor setups without extensive modifications for inert atmosphere handling. Moreover, the avoidance of toxic heavy metal catalysts means that the downstream purification process is vastly simplified, reducing the reliance on expensive scavengers or complex chromatographic separations. This method not only widens the applicability of trifluoromethyl triazole synthesis but also provides a safer, more environmentally benign pathway that aligns perfectly with the principles of green chemistry and sustainable manufacturing practices demanded by modern supply chains.
Mechanistic Insights into Iodine-Promoted Cyclization
To fully appreciate the technical superiority of this method, one must delve into the mechanistic intricacies that drive the formation of the 1,2,4-triazole ring. The reaction is believed to initiate with a base-promoted intermolecular carbon-nitrogen bond formation between the trifluoroethylimidoyl chloride and the hydrazone substrate. This initial condensation yields a trifluoroacetamidine intermediate, which subsequently undergoes isomerization to position the reactive centers appropriately for cyclization. The introduction of elemental iodine serves a dual purpose: it acts as a mild oxidant to facilitate the oxidative iodination of the intermediate, generating an electrophilic species that is primed for the subsequent ring-closing step. This iodine-mediated activation is crucial, as it lowers the energy barrier for the formation of the N-N bond within the triazole ring, a step that is often kinetically challenging in non-catalyzed systems.
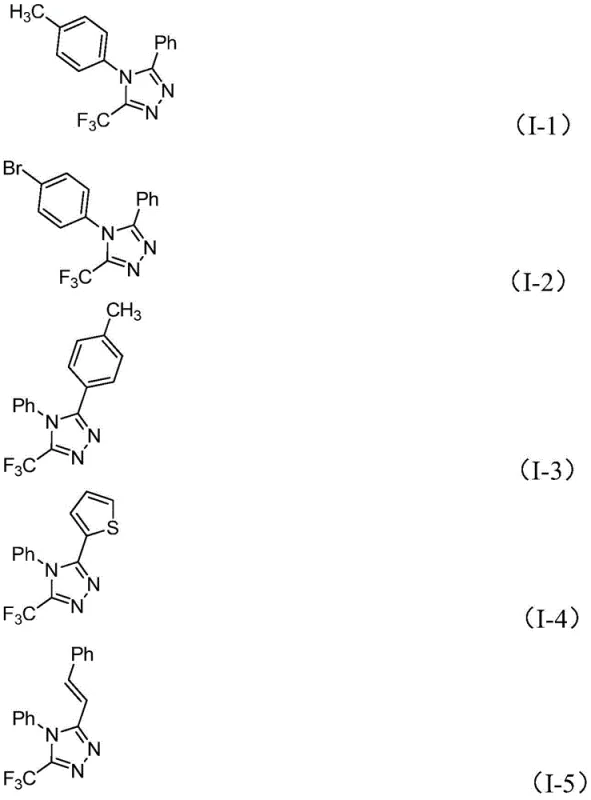
Following the oxidative activation, the system undergoes an intramolecular electrophilic substitution reaction, leading to the closure of the five-membered heterocyclic ring. The final stage involves aromatization, driven by the elimination of hydrogen iodide or related byproducts, to yield the stable 5-trifluoromethyl substituted 1,2,4-triazole product. This mechanistic pathway is highly advantageous for impurity control, as the mild reaction conditions minimize the formation of polymeric byproducts or decomposition species that are common in harsher acidic or basic environments. The tolerance of the system to various functional groups is evidenced by the successful synthesis of diverse derivatives, including those with electron-rich thiophene rings and electron-deficient nitro groups, as shown in the specific examples below. This broad functional group compatibility ensures that the process can be adapted for the synthesis of a wide array of complex analogues required for structure-activity relationship (SAR) studies.
How to Synthesize 5-Trifluoromethyl-1,2,4-Triazoles Efficiently
Implementing this synthesis in a laboratory or pilot plant setting requires adherence to specific procedural parameters to maximize yield and purity. The protocol outlined in the patent provides a robust framework for executing the reaction, emphasizing the importance of reagent stoichiometry and temperature control. Typically, the reaction is initiated by combining sodium acetate, the trifluoroethylimidoyl chloride, and the hydrazone in a suitable organic solvent such as DCE. The mixture is heated to approximately 80°C and maintained for a period of 2 to 4 hours to ensure complete conversion of the starting materials into the amidine intermediate. Following this initial phase, elemental iodine is introduced to the reaction vessel to drive the oxidative cyclization, with the reaction continuing for an additional 1 to 2 hours. Detailed standardized synthesis steps follow below.
- Mix sodium acetate, trifluoroethylimidoyl chloride, and hydrazone in an organic solvent such as dichloroethane (DCE) within a reaction vessel.
- Heat the reaction mixture to 80°C and maintain stirring for 2 to 4 hours to facilitate the initial condensation and cyclization steps.
- Add elemental iodine to the system and continue heating for an additional 1 to 2 hours to promote oxidative aromatization, followed by standard filtration and purification.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this iodine-promoted synthesis method offers tangible strategic advantages that extend far beyond simple chemical yield. The primary value proposition lies in the drastic simplification of the supply chain for raw materials. Unlike methods requiring exotic fluorinating agents or unstable diazo compounds, this process relies on trifluoroethylimidoyl chloride and hydrazones, which are commodity chemicals available from multiple global suppliers. This diversification of the supplier base mitigates the risk of single-source dependency and ensures business continuity even during market fluctuations. Furthermore, the elimination of precious metal catalysts removes a significant cost center associated with both the purchase of the catalyst and the subsequent validation of its removal, directly contributing to cost reduction in API manufacturing.
- Cost Reduction in Manufacturing: The economic impact of switching to this metal-free protocol is profound, primarily driven by the removal of expensive transition metals and the simplification of purification workflows. Traditional heavy metal-catalyzed reactions often require costly scavenging resins or multiple recrystallization steps to meet residual metal specifications, which adds time and material costs to every batch. By utilizing elemental iodine, a cheap and abundant reagent, the process eliminates these downstream burdens, allowing for a more direct isolation of the product. Additionally, the ability to run the reaction under aerobic conditions removes the capital expenditure associated with maintaining inert atmosphere infrastructure, such as nitrogen generators or argon supplies, further lowering the overhead costs per kilogram of produced intermediate.
- Enhanced Supply Chain Reliability: From a logistics perspective, the stability and ease of handling of the reagents involved in this process significantly enhance supply chain reliability. The starting materials are solids or stable liquids that do not require cold chain shipping or special hazard classifications associated with explosive diazo compounds. This ease of transport allows for larger inventory buffers to be held safely, reducing the frequency of urgent orders and minimizing the risk of production stoppages due to material shortages. The robustness of the reaction conditions also means that the process is less sensitive to minor variations in utility quality, such as slight fluctuations in cooling water temperature or steam pressure, ensuring consistent output quality regardless of minor operational variances in different manufacturing sites.
- Scalability and Environmental Compliance: Scaling chemical processes from the bench to the plant floor often reveals hidden complexities, but this method is inherently designed for scalability. The absence of exothermic hazards associated with diazo decomposition and the use of standard solvents like DCE make the heat management of the reaction straightforward in large reactors. Environmentally, the process generates less hazardous waste compared to methods using stoichiometric amounts of heavy metals or toxic fluorinating reagents. The waste stream is easier to treat, and the overall E-factor (mass of waste per mass of product) is improved, aligning with increasingly strict environmental regulations and corporate sustainability goals. This compliance advantage reduces the regulatory burden and potential fines, securing the long-term viability of the manufacturing operation.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These answers are derived directly from the experimental data and claims presented in the patent documentation, providing a clear understanding of the method's capabilities and limitations. Understanding these details is crucial for technical teams evaluating the feasibility of integrating this route into their existing production portfolios. The responses cover aspects ranging from catalyst recovery to substrate flexibility, ensuring a comprehensive overview for decision-makers.
Q: Does this synthesis method require expensive transition metal catalysts?
A: No, the method described in patent CN110467579B utilizes elemental iodine as a promoter rather than expensive heavy metal catalysts like palladium or copper, significantly simplifying downstream purification and reducing heavy metal residue risks in the final API.
Q: What are the reaction conditions regarding moisture and oxygen sensitivity?
A: Unlike many traditional trifluoromethylation protocols that demand strict anhydrous and anaerobic environments, this novel approach operates effectively under ambient atmospheric conditions without the need for rigorous exclusion of moisture or oxygen, lowering operational complexity.
Q: Is the substrate scope limited to specific aromatic groups?
A: The method exhibits broad substrate tolerance, successfully accommodating various substituted aryl groups including those with electron-donating methyl and methoxy groups, as well as electron-withdrawing halogens and nitro groups, allowing for diverse structural modifications.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 5-Trifluoromethyl-1,2,4-Triazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of robust and scalable synthetic routes in the development of next-generation therapeutics. Our team of expert process chemists has thoroughly analyzed the technology disclosed in CN110467579B and is fully prepared to leverage this iodine-promoted methodology for your specific project needs. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from clinical trials to market launch is seamless and uninterrupted. Our state-of-the-art facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications, guaranteeing that every batch of 5-trifluoromethyl-1,2,4-triazole intermediate we deliver meets the highest industry standards for quality and consistency.
We invite you to collaborate with us to optimize your supply chain and reduce your overall manufacturing costs. By partnering with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements and purity needs. We encourage you to reach out today to discuss your project specifics,索取 specific COA data for our existing catalog of triazole derivatives, and obtain detailed route feasibility assessments for your custom targets. Let us be your trusted partner in delivering high-quality chemical solutions that drive your innovation forward.