Advanced Manufacturing of High-Purity Flurbiprofen Axetil for Global Pharmaceutical Supply Chains
Introduction to Next-Generation Flurbiprofen Axetil Manufacturing
The pharmaceutical industry continuously demands higher purity standards for active pharmaceutical ingredients (APIs) and their intermediates, particularly for parenteral formulations where impurity profiles are strictly regulated. Patent CN103254075A introduces a significant advancement in the preparation of Flurbiprofen Axetil, a potent non-steroidal anti-inflammatory drug (NSAID) prodrug widely used in lipid microsphere injections for targeted pain relief. This proprietary methodology addresses the critical limitation of existing industrial grades, which often fail to meet the stringent ≥99.0% purity threshold required for direct formulation without additional, costly purification cycles. By optimizing the esterification conditions and implementing a robust downstream processing protocol involving specific vacuum distillation parameters, this technology enables the production of ultra-high purity Flurbiprofen Axetil that is immediately suitable for sensitive drug delivery systems.
For R&D directors and procurement specialists, understanding the nuances of this synthesis is vital for securing a reliable pharmaceutical intermediate supplier capable of delivering consistent quality. The process leverages readily available starting materials and avoids the use of hazardous transition metal catalysts, thereby simplifying the regulatory dossier and reducing the environmental footprint of the manufacturing process. This report analyzes the technical merits of this approach, highlighting how it translates into tangible commercial advantages such as reduced lead time for high-purity pharmaceutical intermediates and enhanced supply chain reliability. As we delve into the mechanistic details and operational parameters, it becomes clear that this method represents a paradigm shift towards more efficient and scalable production of complex ester-based therapeutics.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthetic routes for Flurbiprofen Axetil have historically struggled with achieving the requisite purity levels for injectable applications in a single pass. Conventional methods often rely on aggressive reaction conditions or less selective coupling agents that generate a complex matrix of byproducts, including unreacted acids, halogenated impurities, and polymeric residues. These impurities necessitate extensive downstream purification, such as multiple recrystallizations or column chromatography, which drastically reduces overall yield and increases the cost of goods sold (COGS). Furthermore, the presence of trace metallic catalysts from traditional coupling reagents poses a significant risk for parenteral products, requiring expensive scavenging steps to meet ICH Q3D guidelines. The cumulative effect of these inefficiencies is a supply chain that is vulnerable to delays and quality deviations, making it difficult for manufacturers to guarantee the consistent availability of high-purity raw materials needed for large-scale commercial production.
The Novel Approach
In stark contrast, the novel approach detailed in the patent utilizes a highly selective esterification strategy employing bromoethyl acetate as the acylating agent in the presence of mild alkali metal salts. This method operates under moderate thermal conditions, typically between 35°C and 60°C, which inherently suppresses thermal degradation and racemization pathways common in harsher protocols. The innovation lies not just in the reaction itself but in the integrated workup procedure, which combines specific aqueous washes with precision vacuum distillation. By collecting fractions strictly within the 173-175°C range at 0.8mmHg, the process effectively isolates the target molecule from close-boiling impurities. This streamlined workflow eliminates the need for laborious chromatographic separations, resulting in a process that is not only chemically superior but also operationally simpler, offering substantial cost savings in API manufacturing through reduced solvent consumption and shorter cycle times.
Mechanistic Insights into Base-Catalyzed Esterification
The core of this synthesis relies on a nucleophilic substitution mechanism where the carboxylate anion of Flurbiprofen attacks the electrophilic carbon of bromoethyl acetate. The choice of catalyst is paramount; the patent specifies the use of alkali metal organic acid salts like sodium acetate or inorganic salts like sodium phosphate. These mild bases serve a dual function: they deprotonate the carboxylic acid to generate the reactive nucleophile without creating a strongly alkaline environment that could hydrolyze the sensitive ester bonds or induce elimination reactions on the bromoethyl moiety. This delicate balance ensures high conversion rates while maintaining the structural integrity of the biphenyl scaffold. The reaction kinetics are further optimized by selecting polar aprotic solvents such as acetone or ethyl acetate, which stabilize the transition state and facilitate the dissolution of both the organic substrate and the inorganic catalyst, ensuring homogeneous reaction conditions that are critical for reproducibility on a commercial scale.
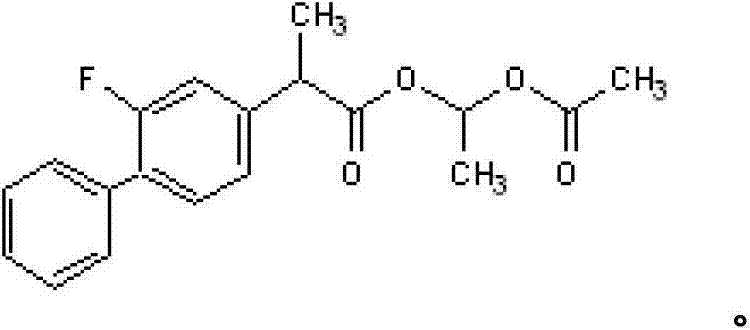
Impurity control is achieved through a multi-stage purification logic embedded within the process design. Following the initial reaction, the crude mixture undergoes a strategic washing sequence using alkali metal carbonate solutions. This step is crucial for neutralizing any residual acidic species and removing water-soluble inorganic salts formed during the reaction. Subsequently, the vacuum distillation step acts as the final polishing stage. Unlike atmospheric distillation, which might expose the thermally sensitive ester to degradation temperatures, reduced pressure distillation allows for separation at significantly lower thermal energy inputs. This preserves the optical purity and chemical stability of the Flurbiprofen Axetil, ensuring that the final product meets the rigorous specifications for single impurity levels (<0.1%) required for clinical applications. The result is a robust process capable of consistently delivering material with purity levels exceeding 99.7%, effectively mitigating the risks associated with batch-to-batch variability.
How to Synthesize Flurbiprofen Axetil Efficiently
The synthesis of this high-value intermediate requires precise adherence to the optimized reaction parameters to ensure maximum yield and purity. The process begins with the careful selection of reagent grades and the maintenance of anhydrous conditions where necessary to prevent premature hydrolysis. Operators must monitor the reaction temperature closely, keeping it within the preferred window of 40°C to 50°C to balance reaction rate with selectivity. Following the reaction completion, the separation of the organic phase must be performed meticulously to avoid emulsion formation, which can trap impurities. The subsequent distillation requires high-vacuum equipment capable of maintaining stable pressure at 0.8mmHg to ensure the accurate collection of the target fraction. For a detailed breakdown of the specific operational steps, reagent ratios, and safety precautions, please refer to the standardized synthesis guide below.
- React flurbiprofen with bromoethyl acetate in the presence of a mild base catalyst like sodium acetate or sodium phosphate within a polar aprotic solvent system.
- Perform a rigorous aqueous workup using alkali metal carbonate solutions to separate the organic phase and remove acidic byproducts effectively.
- Purify the crude oil via vacuum distillation at 173-175°C under 0.8mmHg pressure, followed by decolorization and final solvent removal to yield >99% pure product.
Commercial Advantages for Procurement and Supply Chain Teams
From a strategic procurement perspective, adopting this manufacturing route offers profound benefits that extend beyond simple unit price considerations. The elimination of complex purification steps such as column chromatography significantly reduces the consumption of high-purity solvents and silica gel, which are major cost drivers in fine chemical production. This simplification of the workflow translates directly into a more predictable production schedule, allowing suppliers to respond more agilely to fluctuating market demands. For supply chain heads, the reliance on commodity chemicals like sodium acetate and bromoethyl acetate, rather than exotic or controlled reagents, minimizes the risk of raw material shortages. This accessibility ensures a continuous flow of production, safeguarding against the disruptions that often plague the global supply of specialized pharmaceutical intermediates.
- Cost Reduction in Manufacturing: The process achieves cost efficiency primarily through the simplification of the downstream processing train. By replacing energy-intensive and time-consuming purification methods with a single vacuum distillation step, the overall utility consumption per kilogram of product is drastically lowered. Furthermore, the high selectivity of the reaction minimizes the loss of valuable starting materials to side products, improving the effective mass balance. The absence of transition metal catalysts also removes the need for expensive metal scavenging resins and the associated analytical testing for heavy metals, further streamlining the quality control budget and reducing the total cost of ownership for the manufactured intermediate.
- Enhanced Supply Chain Reliability: The robustness of this synthetic route contributes significantly to supply chain resilience. Because the reaction tolerates a broader range of operating conditions without compromising quality, it is less susceptible to minor variations in raw material quality or environmental factors. This inherent stability reduces the frequency of out-of-specification batches, ensuring a higher rate of successful production runs. Additionally, the use of standard chemical engineering unit operations like liquid-liquid extraction and distillation means that the process can be easily transferred between different manufacturing sites or scaled up without requiring specialized, hard-to-source equipment, thereby diversifying the potential supply base.
- Scalability and Environmental Compliance: Scaling this process from pilot to commercial production is straightforward due to the use of well-understood chemical principles and standard equipment. The waste stream generated is primarily aqueous salt solutions and recoverable organic solvents, which are easier to treat and recycle compared to the hazardous waste associated with heavy metal catalysts or chlorinated coupling agents. This aligns with modern green chemistry initiatives and reduces the environmental compliance burden on the manufacturer. The ability to produce large quantities with a smaller environmental footprint enhances the long-term sustainability of the supply chain, making it a preferred choice for environmentally conscious pharmaceutical partners.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of Flurbiprofen Axetil synthesized via this advanced method. These insights are derived directly from the patent specifications and practical manufacturing experience, aiming to clarify the value proposition for potential partners. Understanding these details is essential for making informed decisions about sourcing strategies and formulation development.
Q: What is the primary advantage of this synthesis method over conventional routes?
A: The primary advantage is the ability to achieve pharmaceutical-grade purity (≥99.0%) directly from the synthesis without requiring complex downstream purification steps, significantly reducing processing time and cost.
Q: Which catalysts are preferred for minimizing side reactions in this process?
A: Mild Lewis base catalysts such as sodium acetate or sodium phosphate are preferred. They effectively promote the esterification reaction while minimizing the risk of racemization or degradation associated with stronger bases.
Q: How does the vacuum distillation step contribute to impurity control?
A: Vacuum distillation at specific parameters (173-175°C/0.8mmHg) allows for the precise separation of the target ester from unreacted starting materials and higher boiling point byproducts, ensuring a narrow impurity profile suitable for injectable formulations.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Flurbiprofen Axetil Supplier
At NINGBO INNO PHARMCHEM, we recognize that the quality of the starting material dictates the success of the final drug product. Our technical team has extensively analyzed the pathway described in CN103254075A and possesses the expertise to implement this high-purity synthesis at an industrial scale. We offer extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with unwavering consistency. Our facilities are equipped with rigorous QC labs and stringent purity specifications that exceed industry standards, guaranteeing that every batch of Flurbiprofen Axetil delivered meets the ≥99.0% purity benchmark required for injectable formulations. We are committed to being a transparent and dependable partner in your drug development journey.
We invite you to engage with our technical procurement team to discuss how this optimized manufacturing route can benefit your specific project requirements. By leveraging our capabilities, you can access a Customized Cost-Saving Analysis tailored to your volume needs, demonstrating exactly how this efficient synthesis can lower your overall COGS. We encourage you to request specific COA data and route feasibility assessments to validate the superiority of our material. Let us collaborate to secure a stable, high-quality supply of this critical pharmaceutical intermediate, ensuring your pipeline moves forward without interruption.