Advanced Synthesis of Flurbiprofen Axetil: Enabling Scalable Production of High-Purity Injectable Intermediates
Advanced Synthesis of Flurbiprofen Axetil: Enabling Scalable Production of High-Purity Injectable Intermediates
The pharmaceutical industry constantly demands higher purity standards for active pharmaceutical ingredients (APIs) and their intermediates, particularly for parenteral formulations where impurity profiles directly impact patient safety. Patent CN103254075B introduces a robust preparation method for flurbiprofen axetil, a potent non-steroidal anti-inflammatory drug (NSAID) prodrug widely used in lipid microsphere injections for cancer and post-surgical pain management. This technology addresses the critical limitation of existing industrial-grade materials, which often fail to meet the stringent ≥99.0% purity threshold required for direct formulation without costly secondary purification. By optimizing the esterification of flurbiprofen with bromoethyl acetate using specific alkali metal salt catalysts, this process delivers a superior impurity profile and consistent quality suitable for global regulatory compliance.
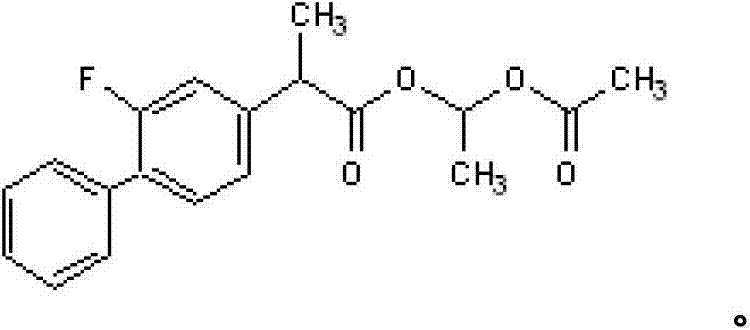
For R&D directors and process chemists, the structural integrity and purity of flurbiprofen axetil are paramount. The molecule, chemically defined as (±) 2-(2-fluoro-4-biphenyl)propionic acid-1-acetoxy ethyl ester, requires precise synthetic control to prevent the formation of toxic by-products or residual halides. The disclosed method utilizes a mild yet effective catalytic system involving sodium acetate or sodium phosphate in solvents such as acetone or ethyl acetate. This approach not only simplifies the reaction setup but also ensures that the resulting crude oil is amenable to high-efficiency purification via vacuum distillation. The ability to achieve purity levels exceeding 99.7% directly from the synthesis line represents a significant technological leap, eliminating the bottleneck of post-synthesis recrystallization or chromatographic purification that typically plagues NSAID intermediate manufacturing.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the production of flurbiprofen axetil for pharmaceutical use has been hindered by the prevalence of industrial-grade starting materials that contain unacceptable levels of impurities. Conventional synthesis routes often rely on harsher conditions or less selective catalysts, leading to complex impurity spectra that include unreacted acids, halogenated by-products, and polymerization residues. These impurities necessitate extensive downstream processing, such as multiple recrystallizations or column chromatography, which drastically reduces overall yield and increases the cost of goods sold (COGS). Furthermore, the variability in purity from batch to batch makes it difficult to standardize the quality of the final injectable lipid microspheres, posing a risk to clinical consistency and regulatory approval. The additional purification steps also extend the manufacturing lead time, creating supply chain vulnerabilities for pharmaceutical companies relying on just-in-time inventory models for their analgesic portfolios.
The Novel Approach
The methodology outlined in CN103254075B overcomes these challenges through a streamlined, high-selectivity esterification process followed by a rigorous physical purification protocol. By employing alkali metal salts like sodium acetate as dual-function catalysts and acid scavengers, the reaction proceeds efficiently at moderate temperatures between 35°C and 60°C, minimizing thermal degradation of the sensitive biphenyl structure. The subsequent work-up involves a strategic washing sequence using aqueous alkali metal carbonates to remove acidic residues, followed by vacuum distillation under inert gas protection. This specific distillation step, collecting fractions at 173-175°C/0.8mmHg, is critical for isolating the target ester with exceptional precision. The result is a colorless, clear oily liquid with a purity of ≥99.0%, often reaching 99.78%, which is directly suitable for formulation, thereby collapsing the traditional multi-step purification timeline into a single, cohesive manufacturing operation.
Mechanistic Insights into Alkali Metal Salt-Catalyzed Esterification
The core of this synthesis lies in the nucleophilic substitution mechanism facilitated by the alkali metal salt catalyst. In the presence of a polar aprotic solvent like acetone or ethyl acetate, the carboxylate anion of flurbiprofen is generated in situ or stabilized by the basic environment provided by the catalyst, such as sodium acetate or disodium hydrogen phosphate. This activated carboxylate species then attacks the electrophilic carbon of bromoethyl acetate, displacing the bromide ion to form the ester linkage. The choice of catalyst is crucial; alkali metal phosphates and acetates provide the optimal balance of basicity to drive the reaction forward without promoting side reactions like elimination or hydrolysis of the ester product. The reaction temperature is carefully controlled between 40°C and 50°C to maximize kinetic energy for the substitution while preventing the thermal decomposition of the bromoethyl acetate reagent, ensuring a molar conversion efficiency that supports yields in the range of 65% to 75%.
Impurity control is achieved through a multi-barrier purification strategy integrated into the workflow. Following the reaction, the mixture is washed with aqueous sodium carbonate solution, which effectively neutralizes and extracts any unreacted flurbiprofen and acidic by-products into the aqueous phase, leaving the neutral ester in the organic oil layer. The subsequent vacuum distillation acts as a molecular sieve, separating the target flurbiprofen axetil from higher boiling point oligomers and lower boiling point solvent residues or unreacted alkylating agents. The use of activated carbon for decolorization further adsorbs trace colored impurities and potential heavy metal contaminants, ensuring the final product meets the visual and chemical specifications required for parenteral use. This comprehensive approach to impurity management ensures that the final API intermediate possesses a clean spectral profile, free from the genotoxic alerts often associated with alkylating agents.
How to Synthesize Flurbiprofen Axetil Efficiently
Implementing this synthesis route requires strict adherence to the specified reaction parameters to guarantee the high-purity outcome described in the patent. The process begins with the dissolution of flurbiprofen in a distilled solvent, followed by the addition of the catalyst and the controlled dropwise addition of bromoethyl acetate to manage exothermicity. Maintaining the reaction temperature within the 35°C-60°C window for a duration of 3 to 7 hours is essential for complete conversion. Post-reaction processing involves phase separation, drying with anhydrous magnesium sulfate, and the critical vacuum distillation step under nitrogen or argon protection to prevent oxidation. The detailed standardized operating procedures for scaling this reaction from laboratory to commercial production are outlined below, providing a clear roadmap for process engineers to replicate the high-yield results.
- React flurbiprofen with bromoethyl acetate in the presence of an alkali metal salt catalyst (e.g., sodium acetate) and a solvent like acetone at 35-60°C.
- Wash the reaction mixture with aqueous alkali metal carbonate to separate the organic oil phase, removing acidic impurities and catalyst residues.
- Purify the crude oil via vacuum distillation collecting fractions at 173-175°C/0.8mmHg, followed by decolorization and solvent removal to yield ≥99.0% pure product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this patented synthesis method offers transformative economic and operational benefits. The primary advantage lies in the drastic simplification of the manufacturing workflow, which eliminates the need for expensive and time-consuming secondary purification steps traditionally required to upgrade industrial-grade material to pharmaceutical standards. By achieving ≥99.0% purity directly from the synthesis and distillation line, manufacturers can significantly reduce solvent consumption, waste generation, and labor costs associated with repetitive recrystallization processes. This streamlining translates into a more competitive cost structure for the final API intermediate, allowing pharmaceutical companies to optimize their margins on high-volume analgesic products without compromising on the stringent quality requirements mandated by health authorities for injectable drugs.
- Cost Reduction in Manufacturing: The elimination of downstream purification bottlenecks directly lowers the operational expenditure per kilogram of produced flurbiprofen axetil. By removing the necessity for additional chromatographic or recrystallization steps, the process reduces solvent usage and energy consumption, leading to substantial cost savings in utility and waste disposal. Furthermore, the use of commercially available and inexpensive catalysts like sodium acetate ensures that raw material costs remain stable and predictable, shielding the supply chain from volatility associated with exotic or proprietary catalytic systems.
- Enhanced Supply Chain Reliability: The robustness of this synthetic route enhances supply continuity by reducing the complexity of the production schedule. With fewer unit operations and a shorter overall cycle time, manufacturers can respond more agilely to fluctuations in market demand for pain management therapies. The reliance on standard chemical reagents and common solvents like acetone and n-hexane ensures that raw material sourcing is not constrained by single-supplier dependencies, thereby mitigating the risk of production stoppages due to raw material shortages and ensuring a steady flow of high-quality intermediates to formulation plants.
- Scalability and Environmental Compliance: The process is inherently designed for scalability, utilizing standard reactor configurations and distillation setups that are easily transferable from pilot plant to multi-ton commercial production. The efficient recovery of solvents through distillation and the minimization of hazardous waste streams align with modern green chemistry principles and environmental regulations. This compliance reduces the regulatory burden on manufacturing sites and facilitates smoother audits, ensuring that the production of this critical pharmaceutical intermediate remains sustainable and uninterrupted in the long term.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of flurbiprofen axetil synthesized via this advanced method. These insights are derived directly from the technical specifications and experimental data provided in the patent documentation, offering clarity on process capabilities and product quality. Understanding these details is crucial for stakeholders evaluating the feasibility of integrating this intermediate into their existing pharmaceutical supply chains or developing new generic formulations of lipid microsphere injections.
Q: What is the critical purity level required for flurbiprofen axetil in injectable formulations?
A: According to patent CN103254075B, the purity must reach ≥99.0% to be suitable for direct use in pharmaceutical preparations, particularly lipid microsphere injections, avoiding the need for additional purification steps that increase costs.
Q: Which catalysts are preferred for this esterification process to ensure high yield?
A: The patent specifies alkali metal organic or inorganic acid salts, specifically sodium acetate or sodium phosphate, which act effectively as both catalysts and acid-binding agents during the reaction with bromoethyl acetate.
Q: How does the vacuum distillation step contribute to the final product quality?
A: Vacuum distillation at 173-175°C/0.8mmHg allows for the precise separation of the target ester from unreacted starting materials and by-products under inert gas protection, ensuring thermal stability and high chemical purity.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Flurbiprofen Axetil Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-purity intermediates play in the efficacy and safety of final drug products. Our technical team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory innovation to industrial reality is seamless and efficient. We are committed to delivering flurbiprofen axetil that meets stringent purity specifications, leveraging our rigorous QC labs to verify every batch against the highest international standards. Our capability to replicate and optimize complex esterification and purification processes allows us to serve as a dependable partner for global pharmaceutical companies seeking to secure their supply of critical analgesic intermediates.
We invite you to engage with our technical procurement team to discuss how our manufacturing capabilities can support your specific project requirements. Whether you need a Customized Cost-Saving Analysis for your current supply chain or require specific COA data and route feasibility assessments for new product development, we are ready to provide the data-driven insights you need. Contact us today to request samples and technical documentation, and let us demonstrate how our commitment to quality and efficiency can drive value for your organization.