Advanced One-Pot Synthesis of 2-Trifluoromethyl Quinazolinones for Commercial Scale-up
Advanced One-Pot Synthesis of 2-Trifluoromethyl Quinazolinones for Commercial Scale-up
The pharmaceutical industry continuously seeks robust synthetic routes for privileged scaffolds that offer enhanced metabolic stability and bioavailability. Patent CN112480015B introduces a groundbreaking multi-component one-pot method for synthesizing 2-trifluoromethyl substituted quinazolinones, a core structure prevalent in numerous bioactive molecules. As illustrated in the structural diversity of known drugs below, the quinazolinone motif is critical for antifungal, antiviral, and anticancer activities. The strategic incorporation of the trifluoromethyl group further amplifies these properties by improving lipophilicity and metabolic resistance, making this synthetic methodology highly valuable for modern drug discovery pipelines.
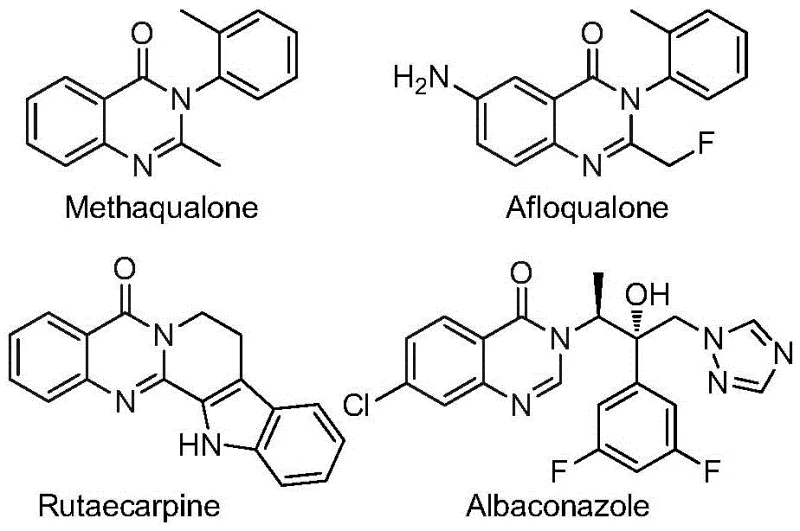
This novel approach addresses the critical need for reliable pharmaceutical intermediate suppliers who can deliver complex heterocycles efficiently. By leveraging a palladium-catalyzed carbonylation cascade, the process transforms inexpensive nitro compounds and trifluoroethylimidoyl chlorides into high-value targets. For R&D directors focused on purity and impurity profiles, this method offers a streamlined pathway that minimizes side reactions often associated with multi-step sequences. The ability to generate these scaffolds in a single operational step represents a significant leap forward in process chemistry, aligning perfectly with the demands for cost reduction in API manufacturing and accelerated timeline-to-market for new therapeutic candidates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of the quinazolinone core has relied on methodologies that present substantial logistical and safety challenges for commercial production. Traditional routes often necessitate the use of high-pressure carbon monoxide gas, which requires specialized autoclave equipment and rigorous safety protocols, thereby inflating capital expenditure and operational complexity. Furthermore, many existing protocols depend on expensive ruthenium or platinum catalysts, or require pre-activated substrates such as 2-bromoformylaniline, which are not only costly but also generate significant halogenated waste streams. These conventional methods frequently suffer from narrow substrate scope and moderate yields, limiting their utility in the rapid generation of diverse analog libraries required for structure-activity relationship (SAR) studies in early-stage drug development.
The Novel Approach
In stark contrast, the methodology disclosed in CN112480015B utilizes a transition metal palladium-catalyzed carbonylation cascade that operates under significantly milder and safer conditions. By employing molybdenum hexacarbonyl (Mo(CO)6) as a solid carbon monoxide surrogate, the process completely eliminates the hazards associated with handling high-pressure CO gas. The reaction proceeds in a true one-pot fashion, combining trifluoroethylimidoyl chloride and nitro compounds directly, as shown in the general reaction scheme below. This telescoped approach not only reduces the number of unit operations but also enhances overall atom economy. The use of cheap and readily available nitro compounds as starting materials drastically lowers the raw material cost base, while the broad functional group tolerance ensures that complex, substituted derivatives can be accessed without extensive protecting group manipulations.
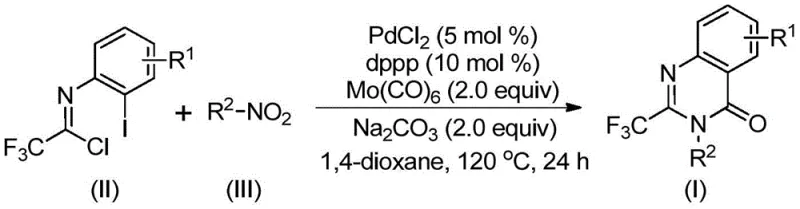
Mechanistic Insights into Pd-Catalyzed Carbonylation Cascade
The mechanistic pathway of this transformation is a sophisticated interplay of reduction, coupling, and cyclization events orchestrated by the palladium catalyst system. Initially, the molybdenum hexacarbonyl serves a dual purpose: it acts as the source of carbon monoxide and facilitates the reduction of the nitro group to the corresponding amine in situ. Once the amine is generated, it undergoes a base-promoted intermolecular carbon-nitrogen bond coupling with the trifluoroethylimidoyl chloride to form a trifluoroacetamidine intermediate. Subsequently, the palladium catalyst, specifically PdCl2 coordinated with the dppp ligand, inserts into the carbon-iodine bond of the substrate. The released carbon monoxide then inserts into the carbon-palladium bond to generate a reactive acyl-palladium species.
This acyl-palladium intermediate is pivotal for the final ring closure. Under the influence of the base, a palladium-nitrogen bond forms, creating a seven-membered cyclic palladium intermediate. The cycle concludes with a reductive elimination step that releases the desired 2-trifluoromethyl substituted quinazolinone product and regenerates the active palladium catalyst. This intricate mechanism ensures high selectivity and minimizes the formation of byproducts, which is crucial for maintaining stringent purity specifications in pharmaceutical intermediates. The choice of 1,3-bis(diphenylphosphino)propane (dppp) as the ligand is particularly effective in stabilizing the palladium center throughout this multi-step cascade, ensuring consistent performance across a wide range of substrates.
How to Synthesize 2-Trifluoromethyl Quinazolinones Efficiently
The execution of this synthesis is designed for operational simplicity, making it highly attractive for both laboratory scale-up and industrial implementation. The protocol involves charging a reaction vessel with the palladium catalyst, ligand, base, CO source, and the two primary organic substrates in an aprotic solvent such as 1,4-dioxane. The detailed standardized synthesis steps are provided in the guide below, outlining the precise molar ratios and thermal conditions required to achieve optimal conversion. This straightforward procedure allows chemists to bypass the complexities of handling gaseous reagents while still accessing high-value heterocyclic cores.
- Combine palladium chloride, dppp ligand, sodium carbonate, Mo(CO)6, trifluoroethylimidoyl chloride, and nitro compound in an organic solvent like dioxane.
- Heat the reaction mixture to 120°C and maintain stirring for 16 to 30 hours to allow the carbonylation cascade to proceed.
- Upon completion, filter the mixture, mix with silica gel, and purify via column chromatography to isolate the target quinazolinone.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this synthetic route offers compelling economic and logistical benefits that extend beyond simple yield improvements. The shift from hazardous gaseous reagents to stable solid surrogates fundamentally alters the risk profile of the manufacturing process, potentially lowering insurance costs and simplifying facility requirements. Moreover, the reliance on commodity chemicals like nitro compounds ensures a robust and continuous supply chain, mitigating the risks associated with sourcing specialized, low-volume precursors. This resilience is critical for maintaining production schedules and meeting the demanding delivery timelines of global pharmaceutical clients.
- Cost Reduction in Manufacturing: The elimination of high-pressure equipment and the use of inexpensive starting materials lead to substantial cost savings in the overall production budget. By avoiding expensive noble metal catalysts like ruthenium or platinum in favor of more accessible palladium systems, and by utilizing solid CO sources that do not require specialized gas handling infrastructure, the capital and operational expenditures are significantly reduced. Additionally, the one-pot nature of the reaction minimizes solvent consumption and waste disposal costs, contributing to a leaner and more cost-effective manufacturing process.
- Enhanced Supply Chain Reliability: The starting materials for this reaction, particularly the nitro compounds and trifluoroethylimidoyl chlorides, are widely available in the global chemical market. This abundance ensures that production is not bottlenecked by the scarcity of exotic reagents. The robustness of the reaction conditions also means that the process is less sensitive to minor variations in raw material quality, further stabilizing the supply chain. For partners seeking a reliable pharmaceutical intermediate supplier, this consistency translates to dependable lead times and uninterrupted material flow.
- Scalability and Environmental Compliance: The protocol has been demonstrated to be scalable, with the patent noting successful expansion to gram-level quantities, indicating a clear path toward kilogram and ton-scale production. The use of standard organic solvents and the absence of toxic heavy metal waste streams (beyond standard palladium recovery) simplify the environmental compliance landscape. The simplified workup procedure, involving filtration and standard chromatography, reduces the complexity of downstream processing, making it easier to integrate into existing GMP manufacturing facilities without major retrofitting.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding this synthesis method. These answers are derived directly from the experimental data and technical specifications outlined in the patent documentation, providing clarity on the practical application of this technology for potential licensees and manufacturing partners.
Q: What are the primary advantages of this one-pot synthesis over traditional methods?
A: This method eliminates the need for hazardous high-pressure carbon monoxide gas by using solid Mo(CO)6 as a safe CO surrogate. It also utilizes cheap, readily available nitro compounds instead of expensive pre-activated substrates, significantly simplifying the supply chain and reducing raw material costs.
Q: Is this process suitable for large-scale industrial production?
A: Yes, the patent explicitly states that the method is operationally simple and has been successfully expanded to the gram level. The use of standard organic solvents like dioxane and common heterogeneous workup procedures (filtration and chromatography) indicates strong potential for commercial scale-up in CDMO facilities.
Q: What is the substrate scope for this reaction?
A: The reaction demonstrates excellent functional group tolerance. It accommodates various substituents on the aromatic ring (R1) including halogens, alkyl groups, and trifluoromethyl groups, as well as diverse amines (R2) such as aryl, alkyl, and cycloalkyl groups, allowing for the synthesis of a wide library of analogs.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Quinazolinone Supplier
At NINGBO INNO PHARMCHEM, we recognize the strategic importance of efficient synthetic routes for complex heterocycles in the development of next-generation therapeutics. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that promising laboratory discoveries can be seamlessly transitioned into full-scale manufacturing. We are committed to delivering high-purity pharmaceutical intermediates that meet stringent purity specifications, supported by our rigorous QC labs and state-of-the-art analytical capabilities. Our expertise in palladium-catalyzed transformations allows us to optimize this specific one-pot protocol for maximum efficiency and yield.
We invite you to collaborate with us to leverage this advanced technology for your drug development programs. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific project needs. We are prepared to provide specific COA data and comprehensive route feasibility assessments to demonstrate how our manufacturing capabilities can accelerate your timeline and reduce your overall cost of goods. Let us be your partner in bringing high-quality 2-trifluoromethyl quinazolinone derivatives to the market.