Advanced Manufacturing of Substituted Imidazopyridine Intermediates for Gastric Acid Inhibitors
Introduction to Next-Generation Imidazopyridine Manufacturing
The pharmaceutical industry's relentless pursuit of more effective gastrointestinal therapies has placed substituted imidazopyridine compounds at the forefront of medicinal chemistry research, particularly as potent inhibitors of H+, K+-ATPase. A pivotal advancement in this domain is documented in Chinese Patent CN1452621A, which discloses a revolutionary process for the large-scale preparation of 2,3-dimethylimidazo[1,2-a]pyridine derivatives. This technology addresses critical bottlenecks in traditional synthetic routes by introducing a novel cyclization strategy that utilizes cyclohexanone as a reaction medium. For R&D directors and process chemists, this represents a significant leap forward, transforming a previously sluggish and low-yielding transformation into a robust, high-efficiency protocol. The patent outlines a method wherein a 5,6-diaminopyridine derivative reacts with a 3-halo-2-butanone compound, achieving conversion rates that consistently exceed 95% under optimized conditions. By shifting the paradigm from conventional inert solvents to this specific ketone-based system, the invention not only enhances chemical efficiency but also aligns with the rigorous demands of modern commercial scale-up of complex pharmaceutical intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art methodologies, such as those described in European Patents EP33094 and WO99/55706, have long plagued manufacturing teams with inherent inefficiencies that hinder cost-effective production. These traditional routes typically rely on solvents like acetone, benzene, or N,N-dimethylformamide (DMF) and often require the presence of inorganic or organic bases to drive the reaction forward. The most detrimental aspect of these legacy processes is the excessive reaction time, which can span anywhere from 16 to 84 hours, coupled with elevated reaction temperatures that strain energy resources. Furthermore, the chemical efficiency is notoriously poor, with isolated yields frequently stagnating between 22% and 55%. Such low throughput necessitates larger reactor volumes and increased raw material consumption to meet production targets, directly impacting the bottom line. For a reliable pharmaceutical intermediate supplier, relying on such antiquated methods creates significant supply chain vulnerabilities, including extended lead times and inconsistent batch quality due to the prolonged exposure of sensitive intermediates to harsh thermal conditions.
The Novel Approach
In stark contrast, the innovative process detailed in CN1452621A leverages cyclohexanone to dramatically accelerate the cyclization kinetics while simultaneously boosting product purity. By reacting the formula (2) precursor with a 3-halo-2-butanone compound in this specific solvent system, the reaction time is slashed to a mere 1 to 4 hours at moderate temperatures of 80-100°C. This acceleration is not merely incremental; it represents a fundamental optimization of the reaction coordinate that allows for rapid throughput without compromising structural integrity. The isolated yields routinely surpass 70%, with many examples demonstrating yields approaching 90%, thereby maximizing the utility of every kilogram of starting material. This approach effectively eliminates the need for prolonged heating and complex work-up procedures associated with high-boiling polar aprotic solvents. As illustrated in the general reaction scheme below, the transformation is direct and highly selective, making it an ideal candidate for cost reduction in API manufacturing where efficiency and speed are paramount.
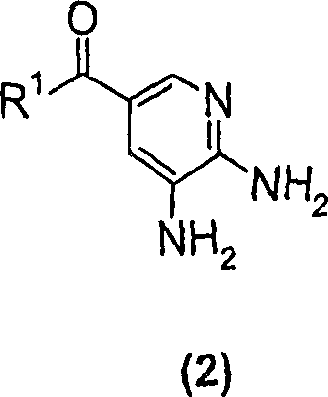
Mechanistic Insights into Cyclohexanone-Mediated Cyclization
The success of this novel methodology lies in the unique solvation properties of cyclohexanone and its interaction with the reactive intermediates formed during the condensation of the diaminopyridine and the alpha-halo ketone. Mechanistically, the reaction proceeds through a nucleophilic attack of the exocyclic amine on the carbonyl carbon of the butanone derivative, followed by an intramolecular cyclization involving the ring nitrogen. The use of cyclohexanone appears to stabilize the transition state or perhaps improve the solubility of the organic salts formed during the progression, preventing the precipitation of intermediates that could otherwise halt the reaction. Furthermore, the patent highlights a sophisticated variation where 3-chloro-2-butanone is used in conjunction with sodium bromide. This facilitates an in-situ Finkelstein-type halogen exchange, generating the more reactive 3-bromo-2-butanone species directly within the reaction mixture. This mechanistic nuance is critical for process chemists, as it allows for the use of cheaper chloro-reagents while maintaining the high reactivity associated with bromo-species. The subsequent reduction of the nitro group in the precursor synthesis, often utilizing a Pd-Ru/C catalyst as shown in the scheme below, ensures that the starting diamino material is of high purity, which is essential for minimizing impurity carry-over into the final cyclized product.
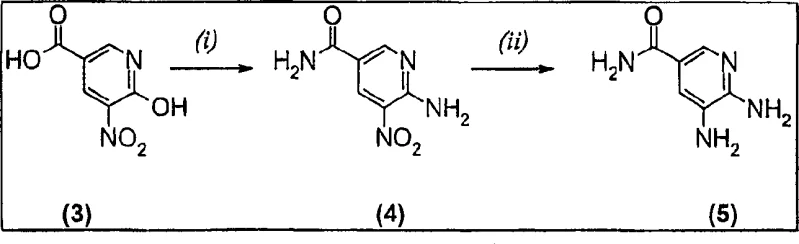
Impurity control is another cornerstone of this mechanistic design. The high conversion rates (>95%) observed indicate that side reactions, such as polymerization of the halo-ketone or over-alkylation, are effectively suppressed under these conditions. The specific stoichiometry, typically employing 1.1 to 2.0 molar equivalents of the halo-ketone, is carefully balanced to drive the reaction to completion without leaving excessive unreacted electrophile that could complicate downstream purification. Additionally, the ability to isolate the product simply by cooling and filtration, as seen in multiple examples, suggests that the product has distinct solubility characteristics in cyclohexanone compared to impurities. This physical property difference is exploited to achieve high-purity pharmaceutical intermediates without the need for resource-intensive chromatographic separations. The robustness of this mechanism is further evidenced by its tolerance to various substituents at the 6-position, whether they are alkoxy groups or amide functionalities, allowing for a versatile platform technology applicable to a wide range of analogues.
![Structure of 8-amino-2,3-dimethylimidazo[1,2-a]pyridine-6-carboxamide demonstrating the amide variant](/insights/img/imidazopyridine-synthesis-pharma-supplier-20260305144027-012.png)
How to Synthesize 8-Amino-2,3-Dimethylimidazopyridine Efficiently
Implementing this synthesis requires precise adherence to the thermal and stoichiometric parameters defined in the patent to ensure reproducibility and safety. The process begins with the suspension of the 5,6-diaminonicotinamide or ester precursor in cyclohexanone, followed by the controlled addition of the halo-ketone reagent. Maintaining the internal temperature between 80°C and 100°C is critical; temperatures below this range may result in incomplete conversion, while significantly higher temperatures could promote degradation. The reaction progress is typically monitored via HPLC, with completion usually achieved within 4 hours. Upon cooling the reaction mixture to ambient or lower temperatures (e.g., 20°C), the product crystallizes out of the solution, allowing for straightforward isolation by filtration. Detailed standard operating procedures regarding specific addition rates, agitation speeds, and drying protocols are essential for scaling this from gram to ton quantities.
- Prepare the 5,6-diaminonicotinate or nicotinamide precursor via nitro reduction using Pd-Ru/C catalyst.
- Suspend the diamino precursor in cyclohexanone and add 3-halo-2-butanone (or generate in-situ using NaBr and 3-chloro-2-butanone).
- Heat the mixture to 80-100°C for 1-4 hours to achieve cyclization, then cool and filter to isolate the high-purity product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this patented process offers tangible strategic benefits that extend far beyond simple chemical yield improvements. The primary advantage lies in the drastic reduction of cycle time; shrinking a reaction from nearly four days to just a few hours effectively multiplies the capacity of existing manufacturing assets without requiring capital expenditure on new reactors. This increased asset turnover directly translates to lower fixed costs per kilogram of product. Moreover, the ability to utilize 3-chloro-2-butanone, a significantly cheaper and more readily available commodity chemical compared to its bromo-counterpart, coupled with inexpensive sodium bromide for the in-situ exchange, results in substantial raw material cost savings. This flexibility in reagent sourcing mitigates supply risk, ensuring that production schedules are not held hostage by the availability of specialized halogenated ketones. The simplified work-up procedure, which often eliminates the need for aqueous extraction and extensive solvent swapping, further reduces utility consumption and waste disposal costs, aligning with modern environmental compliance standards.
- Cost Reduction in Manufacturing: The elimination of long reaction times significantly lowers energy consumption associated with heating and agitation over multi-day periods. Additionally, the high isolated yields (>70% vs historical ~40%) mean that less raw material is wasted, directly improving the cost of goods sold (COGS). The avoidance of expensive transition metal catalysts in the main cyclization step, relying instead on thermal activation in cyclohexanone, removes the need for costly metal scavenging steps often required to meet strict residual metal specifications in pharmaceutical ingredients.
- Enhanced Supply Chain Reliability: By enabling the use of bulk commodity chemicals like 3-chloro-2-butanone and cyclohexanone, the process reduces dependency on niche suppliers of specialized reagents. This diversification of the supply base enhances resilience against market fluctuations and logistical disruptions. The robustness of the reaction, demonstrated by high conversion rates even on multi-kilogram scales, ensures consistent batch-to-batch quality, reducing the risk of production delays caused by failed batches or out-of-specification results that require re-processing.
- Scalability and Environmental Compliance: The process has been successfully demonstrated on a scale of nearly 10 kilograms of starting material, proving its viability for commercial production. The use of cyclohexanone, while requiring appropriate handling, allows for efficient solvent recovery and recycling due to its distinct boiling point and phase behavior. The reduction in reaction time and the simplification of the isolation procedure (filtration vs. extraction) significantly decrease the volume of wastewater generated, facilitating easier compliance with increasingly stringent environmental regulations regarding effluent discharge.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this imidazopyridine synthesis technology. These answers are derived directly from the experimental data and claims presented in the patent literature, providing a factual basis for decision-making. Understanding these nuances is crucial for technical teams evaluating the feasibility of integrating this route into their existing manufacturing portfolios.
Q: What are the primary advantages of using cyclohexanone over traditional solvents like DMF for this synthesis?
A: Cyclohexanone significantly reduces reaction time from up to 84 hours to merely 1-4 hours while increasing isolated yields from roughly 50% to over 70%, drastically improving throughput.
Q: Can the process utilize cheaper chloro-ketones instead of expensive bromo-ketones?
A: Yes, the patent describes an efficient in-situ halogen exchange where sodium bromide and 3-chloro-2-butanone are used to generate the reactive bromo-species, lowering raw material costs.
Q: Is this process suitable for large-scale commercial production of API intermediates?
A: Absolutely. The methodology has been demonstrated on a multi-kilogram scale (e.g., 9.85 kg input) with consistent high conversion rates (>95%) and simplified work-up procedures ideal for manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Imidazopyridine Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in the development of life-saving gastrointestinal medications. Our technical team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory bench to industrial reactor is seamless and efficient. We are committed to delivering products that meet stringent purity specifications, utilizing our rigorous QC labs to verify that every batch conforms to the highest standards required by global regulatory bodies. Our facility is equipped to handle the specific solvent systems and thermal profiles required by the CN1452621A process, guaranteeing a stable and continuous supply of these valuable building blocks for your drug development programs.
We invite you to engage with our technical procurement team to discuss how this advanced manufacturing route can optimize your supply chain. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into the potential economic benefits tailored to your specific volume requirements. We encourage you to contact us today to obtain specific COA data and route feasibility assessments, allowing you to make informed decisions that drive both innovation and profitability in your pharmaceutical projects.