Advanced Metal-Free Synthesis of Trifluoromethyl 1,2,4-Triazines for Commercial Pharmaceutical Manufacturing
The pharmaceutical and agrochemical industries are constantly seeking robust, scalable methodologies for constructing nitrogen-rich heterocyclic scaffolds, particularly those incorporating fluorine atoms to enhance metabolic stability and bioavailability. Patent CN116253692A introduces a groundbreaking preparation method for trifluoromethyl substituted 1,2,4-triazine compounds that addresses many of the historical bottlenecks in this chemical space. This innovation leverages a synergistic [3+3] cycloaddition strategy between chlorohydrazones and trifluoroacetyl sulfur ylides, facilitated by inexpensive inorganic bases. For R&D directors and procurement specialists alike, this represents a significant shift away from transition-metal catalysis towards more sustainable, cost-effective organic synthesis. The ability to generate these complex heterocycles under ambient air conditions without the need for rigorous inert gas protection marks a substantial improvement in operational simplicity and safety profiles for industrial applications.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of the 1,2,4-triazine core has relied heavily on the condensation of amidrazones with 1,2-diketones or alkynes, often requiring elevated temperatures and prolonged reaction times that can degrade sensitive functional groups. Alternative multicomponent reactions involving hydrazides and dicarbonyl compounds frequently suffer from poor atom economy and generate significant amounts of waste, complicating downstream purification processes. Furthermore, many traditional cyclization protocols necessitate the use of stoichiometric amounts of dehydrating agents or harsh acidic conditions, which limit the substrate scope and can lead to the decomposition of acid-sensitive moieties. The reliance on specific, hard-to-synthesize precursors in older methods also restricts the structural diversity accessible to medicinal chemists, often resulting in low overall yields and inconsistent batch-to-batch reproducibility. These inefficiencies translate directly into higher manufacturing costs and longer lead times for active pharmaceutical ingredient (API) development.
The Novel Approach
In stark contrast, the methodology disclosed in the patent utilizes a direct cycloaddition between readily available chlorohydrazones and trifluoroacetyl sulfur ylides, bypassing the need for pre-functionalized diketones. This novel route operates efficiently at room temperature (20-40°C) in common organic solvents like tetrahydrofuran (THF), demonstrating exceptional tolerance for a wide array of functional groups including halogens, ethers, and alkyl chains. 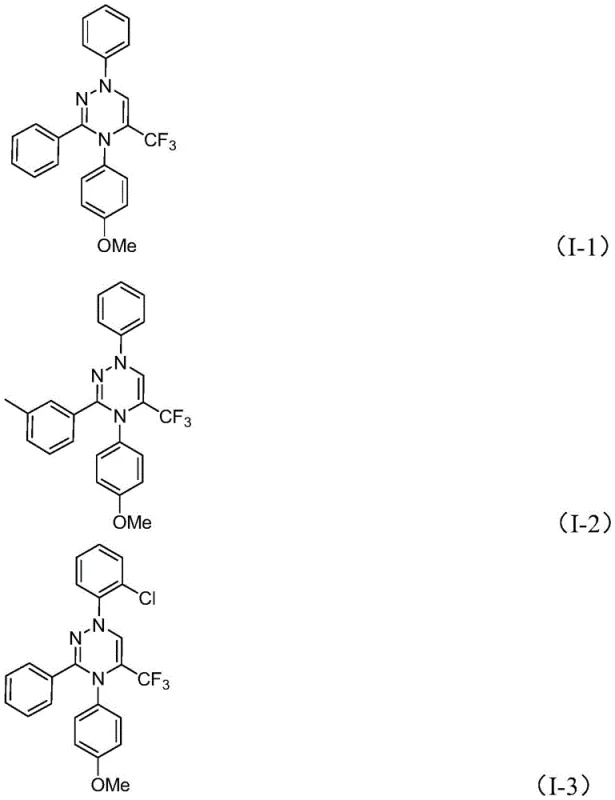 As illustrated by the successful synthesis of diverse derivatives such as compounds I-1 through I-5, this approach offers unparalleled flexibility in modifying the R1, R2, and R3 positions, enabling the rapid generation of focused libraries for structure-activity relationship (SAR) studies. The elimination of heavy metal catalysts not only reduces raw material costs but also removes the stringent requirement for residual metal testing, a critical quality control hurdle in pharmaceutical manufacturing. This streamlined process significantly enhances the feasibility of producing high-purity intermediates on a commercial scale.
As illustrated by the successful synthesis of diverse derivatives such as compounds I-1 through I-5, this approach offers unparalleled flexibility in modifying the R1, R2, and R3 positions, enabling the rapid generation of focused libraries for structure-activity relationship (SAR) studies. The elimination of heavy metal catalysts not only reduces raw material costs but also removes the stringent requirement for residual metal testing, a critical quality control hurdle in pharmaceutical manufacturing. This streamlined process significantly enhances the feasibility of producing high-purity intermediates on a commercial scale.
Mechanistic Insights into Base-Promoted [3+3] Cycloaddition
The core of this transformative synthesis lies in the base-promoted generation of a reactive nitrile imine intermediate, which subsequently undergoes a concerted cycloaddition with the sulfur ylide. Upon addition of potassium carbonate, the chlorohydrazone undergoes dehydrohalogenation to release hydrogen chloride, forming the highly electrophilic nitrile imine species in situ. This intermediate then engages in a synergistic [3+3] cycloaddition reaction with the nucleophilic carbon of the trifluoroacetyl sulfur ylide. 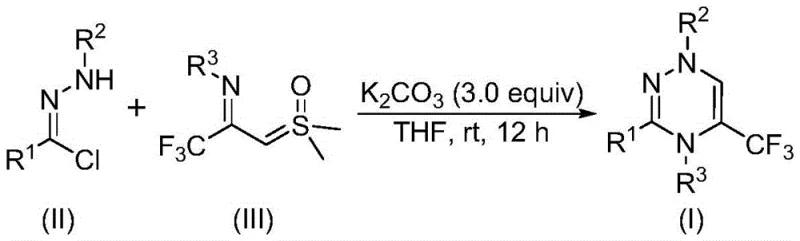 The reaction proceeds through a cyclic transition state that eventually eliminates a molecule of dimethyl sulfoxide (DMSO) to aromatize the ring, yielding the stable 1,2,4-triazine framework. Alternatively, the mechanism may proceed via a stepwise pathway involving intermolecular nucleophilic addition followed by intramolecular nucleophilic substitution, both of which are energetically favorable under the mild reaction conditions employed. This mechanistic elegance ensures high conversion rates and minimizes the formation of polymeric byproducts often seen in radical-based cyclizations.
The reaction proceeds through a cyclic transition state that eventually eliminates a molecule of dimethyl sulfoxide (DMSO) to aromatize the ring, yielding the stable 1,2,4-triazine framework. Alternatively, the mechanism may proceed via a stepwise pathway involving intermolecular nucleophilic addition followed by intramolecular nucleophilic substitution, both of which are energetically favorable under the mild reaction conditions employed. This mechanistic elegance ensures high conversion rates and minimizes the formation of polymeric byproducts often seen in radical-based cyclizations.
From an impurity control perspective, the use of potassium carbonate as a mild, non-nucleophilic base prevents side reactions such as hydrolysis of the trifluoromethyl group or over-alkylation of the nitrogen centers. The reaction's tolerance to air and moisture further mitigates the risk of oxidation byproducts that typically plague sensitive organometallic couplings. By avoiding transition metals like palladium or copper, the process inherently produces a cleaner crude reaction mixture, simplifying the purification workflow to standard silica gel column chromatography. This purity profile is essential for meeting the rigorous specifications required for pharmaceutical intermediates, where trace metal contaminants can catalyze degradation of the final drug product during storage. The robust nature of this mechanism allows for consistent performance across different batches, ensuring reliable supply chain continuity for downstream customers.
How to Synthesize Trifluoromethyl Substituted 1,2,4-Triazine Efficiently
To implement this synthesis effectively, operators should begin by accurately weighing the chlorohydrazone and trifluoroacetyl sulfur ylide substrates according to the optimized molar ratios, typically maintaining a slight excess of the ylide to drive the equilibrium forward. The reaction is initiated by adding three equivalents of anhydrous potassium carbonate to a solution of the substrates in dry tetrahydrofuran, ensuring complete dissolution before stirring commences. Detailed standardized synthesis steps follow below to ensure reproducibility and safety compliance.
- Combine potassium carbonate, chlorohydrazone, and trifluoroacetyl sulfur ylide in an organic solvent such as THF.
- Stir the reaction mixture at room temperature (20-40°C) under an air atmosphere for 10 to 14 hours.
- Filter the mixture, mix with silica gel, and purify via column chromatography to isolate the final trifluoromethyl substituted 1,2,4-triazine compound.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this metal-free synthesis route offers profound strategic benefits that extend far beyond simple yield improvements. The primary advantage lies in the drastic simplification of the supply chain for raw materials; potassium carbonate is a commodity chemical available globally at low cost, unlike specialized ligands or precious metal catalysts which are subject to geopolitical volatility and price fluctuations. By removing the dependency on scarce resources, manufacturers can secure long-term supply contracts with greater confidence, ensuring uninterrupted production schedules even during market disruptions. Furthermore, the elimination of heavy metals removes the need for expensive scavenging resins and complex filtration units, directly lowering the capital expenditure (CAPEX) required for plant setup and the operational expenditure (OPEX) associated with waste disposal and environmental compliance.
- Cost Reduction in Manufacturing: The economic impact of this process is driven by the substitution of expensive catalytic systems with inexpensive inorganic salts, resulting in substantial raw material savings per kilogram of product. Since the reaction proceeds efficiently at room temperature, there is no need for energy-intensive heating or cooling infrastructure, leading to significantly reduced utility costs over the lifecycle of the manufacturing campaign. Additionally, the simplified workup procedure, which avoids complex aqueous extractions often required to remove metal salts, reduces solvent consumption and labor hours, contributing to a leaner and more cost-efficient production model. These cumulative savings allow for more competitive pricing strategies in the global market for pharmaceutical intermediates.
- Enhanced Supply Chain Reliability: The robustness of the reaction conditions, specifically its tolerance to air and ambient moisture, greatly reduces the risk of batch failures due to minor deviations in operating parameters. This resilience translates to higher overall equipment effectiveness (OEE) and more predictable delivery timelines for customers who rely on just-in-time inventory models. The use of stable, shelf-stable starting materials further mitigates the risk of supply interruptions caused by the degradation of sensitive reagents during storage or transport. Consequently, suppliers can offer shorter lead times and more flexible order fulfillment options, strengthening their position as a reliable partner in the pharmaceutical value chain.
- Scalability and Environmental Compliance: Scaling this process from gram to multi-ton quantities is straightforward due to the absence of exothermic hazards associated with strong oxidizers or pyrophoric reagents. The benign nature of the byproducts, primarily dimethyl sulfoxide and inorganic salts, simplifies wastewater treatment and aligns with increasingly stringent environmental regulations regarding heavy metal discharge. This green chemistry profile not only reduces the regulatory burden on manufacturing sites but also enhances the corporate sustainability metrics of the end-product, a factor of growing importance to major pharmaceutical buyers. The ability to scale without re-optimizing critical parameters ensures a seamless transition from pilot plant to full commercial production.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this trifluoromethyl triazine synthesis technology. These answers are derived directly from the experimental data and process descriptions found in the patent literature, providing a factual basis for decision-making. Understanding these nuances is critical for evaluating the feasibility of integrating this route into existing manufacturing portfolios.
Q: What are the key advantages of this trifluoromethyl triazine synthesis method over traditional routes?
A: This method eliminates the need for expensive heavy metal catalysts and operates under mild conditions (room temperature, air atmosphere), significantly simplifying post-treatment and reducing production costs compared to traditional high-temperature condensation reactions.
Q: Is this synthesis method suitable for large-scale commercial production?
A: Yes, the process utilizes cheap and readily available starting materials like potassium carbonate and avoids sensitive reagents, making it highly scalable from gram levels to multi-ton commercial production without complex inert gas protection.
Q: What is the structural diversity achievable with this synthetic route?
A: The method supports a wide range of substituents on the chlorohydrazone and sulfur ylide components, allowing for the synthesis of diverse derivatives including phenyl, naphthyl, and various halogenated or alkyl-substituted 1,2,4-triazine cores.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Trifluoromethyl 1,2,4-Triazine Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of this metal-free cycloaddition technology for the next generation of bioactive molecules. As a premier CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project transitions smoothly from the laboratory bench to the factory floor. Our state-of-the-art facilities are equipped to handle the specific solvent and reagent requirements of this process, while our rigorous QC labs enforce stringent purity specifications to guarantee that every batch meets the highest international standards. We are committed to delivering high-purity pharmaceutical intermediates that empower your drug discovery and development programs.
We invite you to engage with our technical team to explore how this innovative synthesis route can optimize your specific project requirements. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into the economic benefits of switching to this metal-free protocol for your supply chain. Please contact our technical procurement team today to obtain specific COA data for our available trifluoromethyl triazine derivatives and to discuss comprehensive route feasibility assessments tailored to your unique molecular targets.