Advanced One-Pot Synthesis of Carbonyl-Bridged Biheterocyclic Compounds for Pharmaceutical Applications
Advanced One-Pot Synthesis of Carbonyl-Bridged Biheterocyclic Compounds for Pharmaceutical Applications
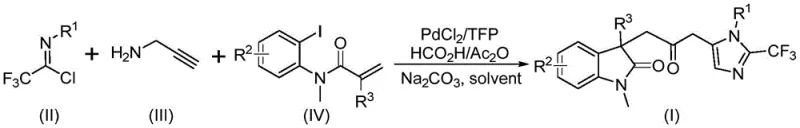
The landscape of organic synthesis for complex heterocyclic scaffolds is constantly evolving, driven by the need for safer, more efficient, and cost-effective methodologies. A significant breakthrough in this domain is detailed in patent CN115353511A, which discloses a novel multi-component method for synthesizing carbonyl-bridged biheterocyclic compounds. This technology addresses critical challenges in constructing indolinone-imidazole hybrid structures, which are prevalent in bioactive molecules and functional materials. By leveraging a transition metal palladium-catalyzed carbonylation cascade, this approach eliminates the need for hazardous carbon monoxide gas while maintaining high reaction efficiency and broad substrate compatibility. For R&D directors and procurement specialists in the pharmaceutical sector, this represents a pivotal shift towards greener and more sustainable manufacturing processes for high-value intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditionally, the construction of carbonyl-bridged biheterocyclic systems has relied on methods that are often fraught with significant operational and safety hurdles. Conventional strategies typically involve the direct coupling of two heterocyclic substrates or oxidative cyclization reactions that require harsh conditions and expensive oxidants. More critically, standard carbonylation reactions frequently necessitate the use of toxic carbon monoxide gas, posing severe safety risks in large-scale manufacturing environments and requiring specialized high-pressure equipment. Furthermore, existing transition metal-catalyzed tandem cyclizations often suffer from limited substrate scope, poor functional group tolerance, and low atom economy, leading to excessive waste generation and increased purification costs. These factors collectively hinder the rapid development and commercial viability of new drug candidates relying on these complex scaffolds.
The Novel Approach
In stark contrast, the methodology described in the patent introduces a streamlined, one-pot synthesis that elegantly circumvents these traditional bottlenecks. By utilizing cheap and readily available starting materials such as trifluoroethylimidoyl chloride, propargylamine, and acrylamide derivatives, the process achieves high efficiency under remarkably mild conditions. The core innovation lies in the use of a formic acid and acetic anhydride mixture as a safe, in situ source of carbon monoxide, effectively removing the dangers associated with handling toxic gas cylinders. This palladium-catalyzed cascade not only simplifies the operational workflow but also demonstrates excellent compatibility with diverse functional groups, allowing for the rapid generation of diversified libraries of trifluoromethyl-substituted biheterocycles essential for modern drug discovery.
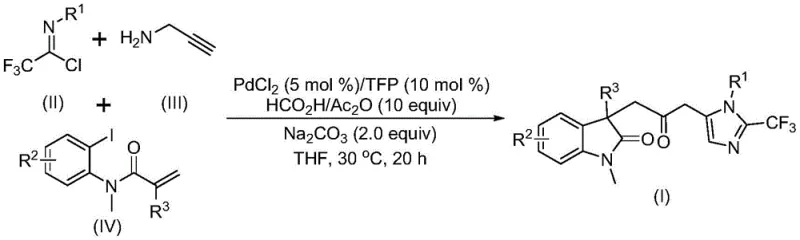
Mechanistic Insights into Palladium-Catalyzed Carbonylation Cascade
The mechanistic pathway of this transformation is a sophisticated sequence of organometallic steps that ensures high chemoselectivity and yield. The reaction is initiated by the insertion of a zero-valent palladium species into the carbon-iodine bond of the acrylamide substrate, followed by an intramolecular Heck reaction to generate a divalent alkyl palladium intermediate. Subsequently, carbon monoxide released from the formic acid and acetic anhydride mixture facilitates a carbonylation event, transforming the alkyl palladium species into a reactive acyl palladium intermediate. Concurrently, a base-promoted intermolecular carbon-nitrogen bond formation occurs between the trifluoroethylimidoyl chloride and propargylamine, yielding a trifluoroacetamidine compound that undergoes isomerization. The cycle concludes with the activation of this amidine by the acyl palladium intermediate, catalyzing an intramolecular cyclization to forge the final carbonyl-bridged biheterocyclic architecture.
From an impurity control perspective, this mechanism offers distinct advantages due to its concerted nature and mild thermal requirements. Operating at a constant temperature of 30°C minimizes the risk of thermal decomposition of sensitive intermediates, which is a common issue in high-temperature cyclizations. The use of specific ligands like trifurylphosphine enhances the stability of the palladium catalyst, reducing the formation of palladium black and associated metal contamination in the final product. Moreover, the stepwise formation of bonds allows for better regulation of side reactions, ensuring that the desired indolinone-imidazole core is formed with high fidelity. This level of control is crucial for pharmaceutical applications where strict purity specifications and well-defined impurity profiles are mandatory for regulatory approval.
How to Synthesize Carbonyl-Bridged Biheterocyclic Compounds Efficiently
Executing this synthesis requires precise attention to reagent stoichiometry and reaction conditions to maximize yield and purity. The general procedure involves charging a reaction vessel with palladium chloride, trifurylphosphine, sodium carbonate, and the formic acid/acetic anhydride mixture in an aprotic organic solvent such as tetrahydrofuran (THF). To this mixture, the three key substrates—trifluoroethylimidoyl chloride, propargylamine, and the specific acrylamide derivative—are added sequentially or simultaneously depending on the specific substrate optimization. The reaction is then stirred at 30°C for a duration of 12 to 20 hours to ensure complete conversion. Post-reaction workup typically involves filtration to remove inorganic salts, followed by silica gel treatment and purification via column chromatography to isolate the target biheterocyclic compound in high purity.
- Combine palladium chloride catalyst, trifurylphosphine ligand, sodium carbonate base, and the formic acid/acetic anhydride CO source mixture in an organic solvent such as THF.
- Add the three key substrates: trifluoroethylimidoyl chloride, propargylamine, and the specific acrylamide derivative into the reaction vessel under stirring.
- Maintain the reaction mixture at 30°C for 12 to 20 hours, followed by filtration, silica gel treatment, and column chromatography purification to isolate the final product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this synthetic route offers tangible benefits that extend beyond mere chemical elegance. The elimination of toxic carbon monoxide gas from the process significantly reduces the regulatory burden and infrastructure costs associated with hazardous material storage and handling. This safety enhancement translates directly into lower insurance premiums and simplified compliance protocols, making the supply chain more resilient and less prone to disruptions caused by safety audits or incidents. Furthermore, the use of commercially available and inexpensive starting materials ensures a stable supply base, mitigating the risks associated with sourcing exotic or custom-synthesized reagents that often plague complex intermediate production.
- Cost Reduction in Manufacturing: The economic viability of this process is underpinned by the use of low-cost catalysts and reagents, coupled with the avoidance of expensive high-pressure equipment required for gaseous CO reactions. By operating at ambient pressure and mild temperatures, energy consumption is drastically minimized, leading to substantial operational expenditure savings. Additionally, the high reaction efficiency and yield reduce the amount of raw material wasted, optimizing the overall cost of goods sold (COGS) for the final pharmaceutical intermediate.
- Enhanced Supply Chain Reliability: The robustness of this method across a wide range of substrates means that production schedules are less likely to be impacted by the need for process re-optimization when switching between different analogues. The scalability of the reaction, demonstrated successfully at the gram scale, provides confidence for seamless transition to kilogram and tonne-level production. This reliability ensures consistent delivery timelines for downstream API manufacturers, fostering stronger long-term partnerships and reducing the lead time for bringing new drugs to market.
- Scalability and Environmental Compliance: From an environmental standpoint, the in situ generation of carbon monoxide aligns with green chemistry principles by reducing the carbon footprint associated with gas transport and storage. The simplified post-treatment process, involving standard filtration and chromatography, generates less hazardous waste compared to traditional multi-step syntheses. This ease of waste management facilitates compliance with increasingly stringent environmental regulations, ensuring uninterrupted production capabilities and enhancing the corporate sustainability profile of the manufacturing entity.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this patented synthesis method. These insights are derived directly from the experimental data and beneficial effects outlined in the patent documentation, providing a clear understanding of the technology's practical application. Understanding these details is essential for stakeholders evaluating the feasibility of integrating this route into their existing manufacturing portfolios or R&D pipelines.
Q: What is the primary safety advantage of this synthesis method compared to traditional carbonylation?
A: Unlike conventional methods that require handling toxic carbon monoxide gas cylinders, this protocol utilizes a formic acid and acetic anhydride mixture to generate carbon monoxide in situ, significantly enhancing operational safety.
Q: What represents the optimal reaction temperature and time for this transformation?
A: The patent data indicates that the reaction proceeds efficiently at a mild temperature of 30°C over a period of 12 to 20 hours, balancing conversion rates with energy consumption.
Q: Is this method scalable for industrial production of pharmaceutical intermediates?
A: Yes, the methodology has been successfully expanded to gram-scale reactions with high yields, demonstrating robust substrate compatibility and potential for commercial scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Carbonyl-Bridged Biheterocyclic Compounds Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of advanced catalytic methodologies like the one described in patent CN115353511A for accelerating drug discovery and development. As a premier CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from benchtop to plant floor is seamless and efficient. Our state-of-the-art facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications, guaranteeing that every batch of carbonyl-bridged biheterocyclic compounds delivered meets the highest industry standards for pharmaceutical intermediates.
We invite you to collaborate with us to leverage this cutting-edge technology for your specific project needs. Our technical team is ready to provide a Customized Cost-Saving Analysis tailored to your volume requirements, demonstrating how this efficient synthesis can optimize your budget without compromising quality. Please contact our technical procurement team today to request specific COA data and comprehensive route feasibility assessments, and let us help you secure a reliable supply of these critical building blocks for your next generation of therapeutics.