Advancing Antiviral Drug Synthesis: A Novel Organocatalytic Route for Chiral Carbocyclic Purine Nucleosides
The pharmaceutical industry continuously seeks more efficient pathways to synthesize complex antiviral agents, particularly chiral carbocyclic purine nucleosides which serve as critical scaffolds for drugs like Abacavir and Entecavir. Patent CN108558882B introduces a groundbreaking methodology that leverages asymmetric [3+2] cycloaddition to construct these vital five-membered carbocyclic structures with high precision. This innovation represents a significant leap forward in organic synthesis, moving away from laborious chiral pool strategies toward a more direct, catalytic approach. By utilizing alpha-purine-substituted acrylates and allenoic acid esters as primary building blocks, the process achieves remarkable stereocontrol without the need for stoichiometric chiral auxiliaries. The ability to generate optically pure intermediates directly from achiral precursors not only simplifies the synthetic route but also opens new avenues for cost-effective manufacturing of high-value antiviral intermediates.
![General reaction scheme for the synthesis of chiral five-membered carbocyclic purine nucleosides via asymmetric [3+2] cycloaddition](/insights/img/chiral-purine-nucleoside-synthesis-catalysis-pharma-supplier-20260303144508-01.webp)
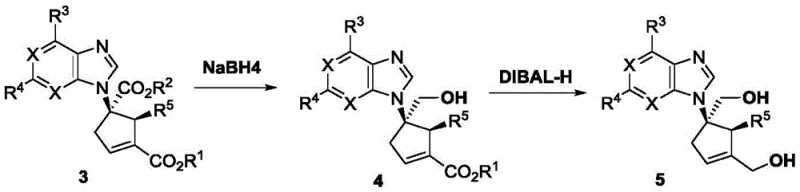
For R&D directors evaluating process feasibility, the mechanistic elegance of this transformation is paramount. The reaction proceeds through a zwitterionic intermediate generated by the nucleophilic attack of the chiral phosphine catalyst on the allenoate. This intermediate then undergoes a highly organized [3+2] cyclization with the electron-deficient acrylate, guided by the steric environment of the catalyst ligand. The patent highlights the use of specialized SITCP-derived ligands, such as C10, which create a rigid chiral pocket essential for differentiating the enantiotopic faces of the reacting species. Furthermore, the resulting cyclic products retain functional handles, specifically ester groups, that allow for downstream diversification. As illustrated in the subsequent derivatization studies, these intermediates can be selectively reduced to mono-alcohols or diols, preserving the established chirality while installing the necessary hydroxyl groups found in final drug molecules.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of chiral five-membered carbocyclic nucleosides has been fraught with synthetic challenges that hinder scalable production. Traditional routes often rely on the chiral pool strategy, where naturally occurring chiral molecules like sugars or amino acids are used as starting materials. While effective for small-scale laboratory synthesis, these methods suffer from inherent limitations when applied to industrial manufacturing. The availability of specific chiral starting materials can be restricted, leading to supply chain bottlenecks and volatile pricing. Moreover, these routes typically require extensive protection and deprotection sequences to manage the multiple functional groups present in the chiral pool precursors. Each additional step introduces potential yield losses and increases the generation of chemical waste, thereby inflating the overall cost of goods. Another common approach involves the resolution of racemic mixtures, which theoretically caps the maximum yield at 50% unless dynamic kinetic resolution is employed, adding further complexity and cost to the process.
The Novel Approach
In stark contrast, the methodology disclosed in Patent CN108558882B offers a streamlined alternative that bypasses many of these historical hurdles. By employing an organocatalytic [3+2] cycloaddition, the synthesis constructs the chiral carbon ring and installs the purine base simultaneously in a single operational step. This convergent strategy drastically reduces the step count compared to linear chiral pool syntheses. The use of simple, achiral acrylates and allenoates as starting materials ensures a robust and reliable supply chain, as these commodities are readily available from multiple chemical vendors. The reaction conditions are notably mild, typically proceeding at temperatures between 0°C and 25°C, which minimizes energy consumption and reduces the risk of thermal degradation of sensitive purine moieties. This efficiency translates directly into improved process mass intensity (PMI), a key metric for green chemistry and sustainable manufacturing, making it an attractive option for companies aiming to reduce their environmental footprint while maintaining high productivity.
Mechanistic Insights into Chiral Phosphine-Catalyzed Cycloaddition
The success of this synthetic route hinges on the precise design of the chiral phosphine catalyst, which acts as the molecular director of stereochemistry. The catalyst, specifically derivatives of the SITCP scaffold like C10, functions by activating the allenoate through the formation of a phosphonium zwitterion. This activation renders the central carbon of the allenoate nucleophilic, allowing it to attack the beta-position of the alpha-purine acrylate. The bulky aryl groups on the phosphine ligand create a defined chiral environment that shields one face of the reacting complex, forcing the bond formation to occur from the opposite, less hindered direction. This steric differentiation is critical for achieving the high enantiomeric excess values reported, often exceeding 90% ee. The mechanism avoids the use of transition metals, which is a significant advantage for pharmaceutical applications where residual metal contamination is strictly regulated. By relying on organocatalysis, the process eliminates the need for expensive metal scavengers and rigorous purification steps designed to remove trace metals, thereby simplifying the downstream processing workflow.
Impurity control is another critical aspect where this mechanism excels. The high regioselectivity of the [3+2] cycloaddition ensures that the purine base is attached at the correct position on the carbocyclic ring, minimizing the formation of regioisomers that are difficult to separate. Furthermore, the mild reaction conditions prevent side reactions such as polymerization of the allenoate or decomposition of the purine ring, which can occur under harsher acidic or basic conditions used in other methods. The patent data indicates that the reaction tolerates a variety of substituents on the purine ring, including chloro, methoxy, and amino groups, without significant loss of selectivity. This substrate scope suggests that the catalytic cycle is robust and adaptable, capable of producing a diverse library of nucleoside analogs. For process chemists, this means that a single optimized protocol can potentially be applied to multiple targets within a drug discovery pipeline, accelerating the timeline from lead optimization to clinical supply.
How to Synthesize Chiral Five-Membered Carbocyclic Purine Nucleosides Efficiently
Implementing this synthesis requires careful attention to reaction parameters to maximize yield and optical purity. The standard protocol involves dissolving the alpha-purine acrylate and allenoate ester in an anhydrous solvent such as dichloromethane under an inert nitrogen atmosphere. The chiral catalyst and an additive, typically 2-naphthol, are added to the mixture, which is then cooled to 0°C to initiate the reaction. Monitoring via thin-layer chromatography (TLC) is essential to determine the optimal endpoint, usually ranging from 10 to 18 hours depending on the specific substrate reactivity. Upon completion, the reaction is quenched and the product is isolated through standard aqueous workup and column chromatography. For detailed operational parameters and safety considerations, please refer to the standardized synthesis guide below.
- Prepare the reaction mixture by combining alpha-purine-substituted acrylate and allenoate ester in a suitable solvent like dichloromethane under inert atmosphere.
- Add the chiral phosphine catalyst (e.g., SITCP derivative) and an additive like 2-naphthol, maintaining the temperature between 0°C and 25°C.
- Monitor the reaction progress via TLC, then perform workup involving extraction, drying, and column chromatography to isolate the chiral product with high enantiomeric excess.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement and supply chain perspective, this technology offers compelling advantages that align with strategic goals of cost reduction and supply security. The shift from complex chiral starting materials to simple achiral commodity chemicals fundamentally alters the cost structure of the synthesis. By removing the dependency on scarce natural chiral sources, manufacturers can mitigate the risks associated with agricultural variability or geopolitical supply disruptions. The simplified synthetic route also implies a reduction in the total volume of solvents and reagents required per kilogram of product, which directly lowers waste disposal costs and raw material expenditure. Additionally, the absence of heavy metal catalysts simplifies the regulatory compliance landscape, reducing the analytical burden and time required for batch release testing. These factors collectively contribute to a more resilient and economically viable supply chain for critical antiviral intermediates.
- Cost Reduction in Manufacturing: The elimination of stoichiometric chiral reagents and the reduction in synthetic step count lead to substantial cost savings. Traditional methods often involve costly resolution steps or expensive chiral auxiliaries that are discarded after use, representing a significant financial inefficiency. In contrast, the catalytic nature of this process means that a small amount of chiral catalyst can produce a large quantity of product, dramatically improving the atom economy. Furthermore, the mild reaction conditions reduce energy consumption for heating or cooling, contributing to lower utility costs. The simplified purification process, free from metal removal steps, also reduces the consumption of specialized resins and filtration media, further driving down the operational expenses associated with large-scale production.
- Enhanced Supply Chain Reliability: Sourcing reliability is significantly improved by the use of widely available starting materials. Alpha-purine acrylates and allenoates are standard building blocks in organic synthesis, available from a broad network of global chemical suppliers. This diversity of supply sources prevents single-point failures in the supply chain, ensuring continuous production even if one vendor faces disruptions. The robustness of the reaction conditions also means that the process is less sensitive to minor variations in raw material quality, reducing the rate of batch failures. For supply chain managers, this translates to more predictable lead times and the ability to maintain lower safety stock levels, freeing up working capital and improving overall inventory turnover rates.
- Scalability and Environmental Compliance: The process is inherently scalable due to its exothermic profile and lack of hazardous reagents. The reaction can be safely conducted in standard glass-lined or stainless steel reactors without the need for specialized high-pressure or cryogenic equipment. This ease of scale-up facilitates rapid technology transfer from laboratory to pilot plant and finally to commercial manufacturing. From an environmental standpoint, the organocatalytic approach aligns with green chemistry principles by avoiding toxic heavy metals and reducing solvent usage through higher concentration reactions. This compliance with increasingly stringent environmental regulations minimizes the risk of regulatory penalties and enhances the corporate sustainability profile, which is becoming a key differentiator in the global pharmaceutical market.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding this synthesis method. They are derived from the specific experimental data and beneficial effects outlined in the patent documentation. Understanding these details helps stakeholders assess the feasibility of integrating this technology into their existing manufacturing portfolios. The answers reflect the current state of the art as described in the intellectual property, providing a factual basis for decision-making.
Q: What are the key advantages of this [3+2] cycloaddition method over traditional chiral pool synthesis?
A: This method utilizes readily available achiral raw materials and a chiral organocatalyst to construct the stereocenter in a single step, eliminating the need for expensive chiral starting materials and complex multi-step resolutions required in traditional approaches.
Q: What level of enantioselectivity can be achieved with this process?
A: The patent data demonstrates excellent stereocontrol, with enantiomeric excess (ee) values reaching up to 96% for various substrates when optimized catalysts like C10 are employed under mild conditions.
Q: Can the resulting cyclic nucleoside intermediates be further functionalized?
A: Yes, the synthesized five-membered carbocyclic nucleosides possess ester groups that can be selectively reduced to mono-alcohols or diols using reagents like NaBH4 or DIBAL-H, providing versatile scaffolds for final API synthesis.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Chiral Carbocyclic Purine Nucleoside Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of this asymmetric [3+2] cycloaddition technology for the production of next-generation antiviral intermediates. As a leading CDMO partner, we possess the technical expertise and infrastructure to translate this innovative patent into commercial reality. Our team has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project moves seamlessly from gram-scale optimization to full-scale manufacturing. We are committed to delivering high-purity pharmaceutical intermediates that meet the most stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation. Our dedication to quality assurance guarantees that every batch conforms to the exacting standards required by global regulatory authorities.
We invite you to collaborate with us to leverage this advanced synthetic route for your specific drug development programs. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your volume requirements and timeline. By partnering with us, you gain access to our deep process knowledge and supply chain network, enabling you to accelerate your time-to-market. Please contact us today to request specific COA data and route feasibility assessments, and let us demonstrate how our manufacturing capabilities can drive value and efficiency for your organization.