Scalable Metal-Free Synthesis of 3,4,5-Trisubstituted 1,2,4-Triazoles for Advanced Pharmaceutical Applications
Scalable Metal-Free Synthesis of 3,4,5-Trisubstituted 1,2,4-Triazoles for Advanced Pharmaceutical Applications
The landscape of heterocyclic chemistry is continuously evolving to meet the rigorous demands of modern drug discovery, particularly for scaffolds that enhance metabolic stability and bioavailability. A pivotal advancement in this domain is detailed in patent CN113105402B, which discloses a highly efficient preparation method for 3,4,5-trisubstituted 1,2,4-triazole compounds. These nitrogen-containing five-membered heterocycles are not merely academic curiosities; they form the critical molecular backbone of several blockbuster pharmaceuticals, including Maraviroc, Sitagliptin, and Deferasirox, as illustrated in the structural diversity shown below. The introduction of a trifluoromethyl group into these heterocyclic systems is known to drastically improve physicochemical properties such as lipophilicity and electronegativity, making this specific synthetic methodology a cornerstone for developing next-generation therapeutic agents.
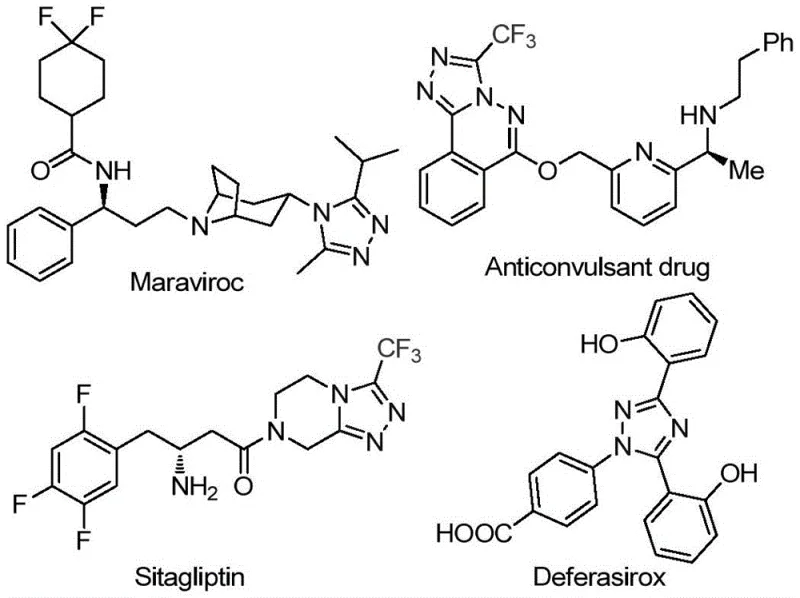
The significance of this patent extends beyond mere structural novelty; it addresses the fundamental challenges of process chemistry. Traditional routes to functionalized triazoles often suffer from reliance on expensive transition metal catalysts, stringent reaction conditions requiring inert atmospheres, and complex workup procedures to remove toxic metal residues. In contrast, the methodology described in CN113105402B leverages a non-metallic iodine-promoted system that operates under relatively mild conditions. This shift represents a paradigm change for procurement and supply chain managers, as it decouples production from the volatility of precious metal markets and simplifies regulatory compliance regarding heavy metal impurities in Active Pharmaceutical Ingredients (APIs).
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of polysubstituted 1,2,4-triazoles, especially those bearing both trifluoromethyl and acyl groups, has been fraught with synthetic inefficiencies. Conventional literature methods frequently employ transition metal catalysts such as copper or palladium to facilitate cyclization, which introduces significant downstream processing burdens. The removal of trace metal residues to meet International Council for Harmonisation (ICH) guidelines often requires specialized scavenging resins or multiple recrystallization steps, driving up both the Cost of Goods Sold (COGS) and the environmental footprint of the manufacturing process. Furthermore, many established protocols necessitate strictly anhydrous and oxygen-free conditions, demanding expensive Schlenk line techniques or glovebox operations that are difficult to translate to multi-tonne commercial reactors. These factors collectively create bottlenecks in the supply chain, increasing lead times and limiting the scalability of potential drug candidates.
The Novel Approach
The innovative strategy outlined in the patent circumvents these obstacles by utilizing a metal-free, iodine-mediated oxidative cyclization. By employing dimethyl sulfoxide (DMSO) not just as a solvent but as an oxygen source in conjunction with elemental iodine, the process achieves a Kornblum oxidation of aryl ethyl ketones in situ. This generates reactive aryl diketone intermediates which subsequently undergo tandem condensation and cyclization with trifluoroethylimide hydrazides. The beauty of this approach lies in its operational simplicity: the reaction tolerates ambient atmosphere, utilizes cheap and readily available reagents, and avoids the generation of hazardous heavy metal waste. For a reliable pharmaceutical intermediate supplier, this translates to a robust process that can be executed in standard glass-lined reactors without the need for exotic infrastructure, thereby enhancing overall production agility and cost-effectiveness.
Mechanistic Insights into Iodine-Promoted Oxidative Cyclization
To fully appreciate the technical merit of this synthesis, one must delve into the mechanistic pathway that drives the formation of the triazole ring. The reaction initiates with the iodination of the aryl ethyl ketone, followed by a Kornblum oxidation where DMSO acts as the oxidant to convert the methyl ketone moiety into an alpha-dicarbonyl species. This electrophilic intermediate is then trapped by the nucleophilic nitrogen of the trifluoroethylimide hydrazide. The subsequent dehydration leads to a hydrazone intermediate, which is poised for the final ring-closing step. Under the continued influence of iodine and the basic environment provided by pyridine and sodium dihydrogen phosphate, an intramolecular cyclization occurs. This sequence effectively constructs the 1,2,4-triazole core while simultaneously installing the crucial trifluoromethyl group at the 3-position and the acyl group at the 5-position, as depicted in the general reaction scheme below.
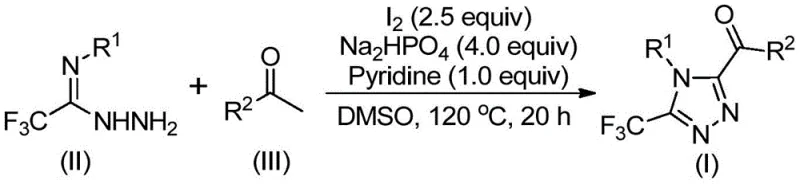
From an impurity control perspective, this mechanism offers distinct advantages. The use of stoichiometric iodine and phosphate buffers helps maintain a controlled pH and oxidation potential, minimizing the formation of over-oxidized byproducts or polymeric tars that often plague radical-mediated reactions. The specificity of the hydrazone condensation ensures that the regioselectivity of the triazole formation is high, reducing the burden on purification teams. Moreover, the tolerance for various substituents on both the aryl ketone (R2) and the hydrazide (R1)—including electron-donating groups like methoxy and electron-withdrawing groups like chloro or trifluoromethyl—demonstrates the versatility of this catalytic system. This broad substrate scope allows medicinal chemists to rapidly generate diverse libraries of analogs for Structure-Activity Relationship (SAR) studies without needing to re-optimize reaction conditions for each new derivative.
How to Synthesize 3,4,5-Trisubstituted 1,2,4-Triazole Efficiently
Implementing this synthesis in a laboratory or pilot plant setting requires adherence to specific thermal and stoichiometric parameters to maximize yield and purity. The process is designed as a sequential one-pot reaction, which minimizes material handling and solvent usage. Initially, the aryl ethyl ketone is activated with iodine in DMSO at moderate temperatures before the introduction of the nitrogen source. This staged addition is critical to ensure the complete formation of the dicarbonyl intermediate prior to cyclization. The detailed standardized synthesis steps, including precise molar ratios and temperature profiles derived from the patent examples, are provided in the guide below for immediate technical reference.
- Mix aryl ethyl ketone and elemental iodine in DMSO, heating to 90-110°C for 4-6 hours to initiate Kornblum oxidation.
- Add additional iodine, sodium dihydrogen phosphate, pyridine, and trifluoroethylimide hydrazide to the reaction mixture.
- Heat the solution to 110-130°C for 12-20 hours to complete the cyclization, then filter and purify via column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For stakeholders focused on the bottom line and supply continuity, the economic implications of this patent are profound. The shift away from precious metal catalysis to a base-metal-free iodine system fundamentally alters the cost structure of manufacturing these high-value intermediates. By eliminating the need for expensive ligands and catalysts, the direct material costs are significantly reduced. Furthermore, the simplified workup procedure—often requiring only filtration and standard column chromatography or recrystallization—drastically cuts down on processing time and solvent consumption. This efficiency gain is not merely theoretical; it directly correlates to higher throughput and lower energy expenditure per kilogram of product, making the commercial scale-up of complex pharmaceutical intermediates much more financially viable.
- Cost Reduction in Manufacturing: The elimination of heavy metal catalysts removes the necessity for costly metal scavenging steps and extensive analytical testing for residual metals, which are major cost drivers in API production. Additionally, the use of commodity chemicals like elemental iodine and DMSO, which are available in bulk quantities at stable prices, insulates the production budget from the volatility associated with rare earth or precious metal markets. The high atom economy of the tandem reaction further ensures that raw material utilization is optimized, minimizing waste disposal costs.
- Enhanced Supply Chain Reliability: The starting materials, specifically aryl ethyl ketones and trifluoroethylimide hydrazides, are commercially available from multiple global vendors, reducing the risk of single-source supply disruptions. The robustness of the reaction conditions, which do not require specialized inert gas lines or ultra-dry solvents, means that production can be maintained even in facilities with standard utility infrastructure. This resilience is crucial for maintaining consistent delivery schedules to downstream pharmaceutical clients who rely on Just-In-Time inventory models.
- Scalability and Environmental Compliance: The process is inherently scalable, as demonstrated by its successful expansion to gram levels in the patent examples without loss of efficiency. The absence of toxic heavy metals simplifies the environmental, health, and safety (EHS) profile of the manufacturing process, facilitating easier permitting and compliance with increasingly stringent global environmental regulations. The use of DMSO, a high-boiling polar aprotic solvent, also allows for effective heat management in large-scale exothermic reactions, ensuring safe operation at the multi-tonne scale.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthetic technology. These insights are derived directly from the experimental data and beneficial effects reported in the patent documentation, providing a clear understanding of the method's capabilities and limitations for potential adopters.
Q: What are the primary advantages of this iodine-promoted method over traditional metal-catalyzed routes?
A: This method eliminates the need for toxic heavy metal catalysts, significantly simplifying downstream purification and reducing environmental hazards. It operates under ambient atmospheric conditions without requiring strict anhydrous or oxygen-free environments, lowering operational complexity and equipment costs.
Q: Which key starting materials are required for synthesizing these trifluoromethyl-containing triazoles?
A: The synthesis utilizes commercially available and inexpensive aryl ethyl ketones and trifluoroethylimide hydrazides. The trifluoroethylimide hydrazide itself can be prepared in high yield from trifluoroacetimidoyl chloride and hydrazine hydrate, ensuring a robust and accessible supply chain for raw materials.
Q: Is this synthetic route suitable for large-scale industrial production?
A: Yes, the patent explicitly states that the method is easily expandable from gram-level laboratory synthesis to industrial scale. The use of common solvents like DMSO and stable reagents like elemental iodine facilitates safe handling and process intensification in commercial manufacturing settings.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3,4,5-Trisubstituted 1,2,4-Triazole Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition from bench-scale discovery to commercial manufacturing requires a partner with deep technical expertise and robust infrastructure. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the promising chemistry described in CN113105402B can be seamlessly translated into a reliable supply stream. We operate stringent purity specifications and maintain rigorous QC labs equipped with advanced analytical instrumentation to guarantee that every batch of 3,4,5-trisubstituted 1,2,4-triazole meets the exacting standards required for pharmaceutical applications.
We invite you to collaborate with us to leverage this cost-effective and scalable synthetic route for your drug development programs. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements. Please contact us today to request specific COA data and route feasibility assessments, and let us demonstrate how our manufacturing capabilities can accelerate your path to market while optimizing your supply chain economics.