Advanced Metal-Free Synthesis of 3,4,5-Trisubstituted 1,2,4-Triazoles for Scalable Pharmaceutical Production
Introduction to Next-Generation Triazole Synthesis
The pharmaceutical industry continuously seeks robust and scalable methodologies for constructing nitrogen-containing heterocycles, particularly the 1,2,4-triazole scaffold which serves as a critical pharmacophore in numerous blockbuster drugs. As illustrated in the structural diversity of bioactive molecules such as Maraviroc, Sitagliptin, and Deferasirox, the incorporation of trifluoromethyl groups into these heterocyclic systems significantly enhances metabolic stability, lipophilicity, and bioavailability. 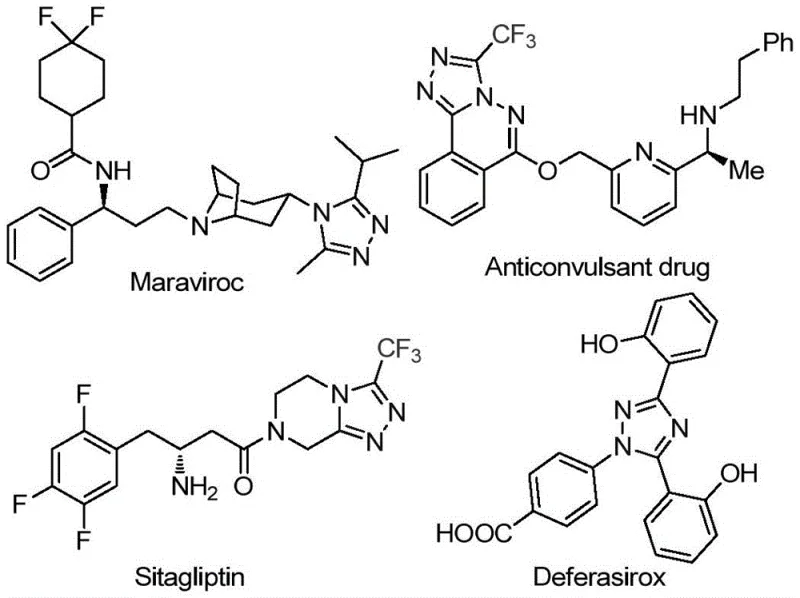 Patent CN113105402B discloses a groundbreaking preparation method for 3,4,5-trisubstituted 1,2,4-triazole compounds that addresses long-standing challenges in synthetic efficiency and environmental compliance. This novel approach leverages a non-metallic iodine-promoted strategy, eliminating the reliance on costly transition metal catalysts while maintaining high functional group tolerance. For R&D directors and procurement specialists, this technology represents a pivotal shift towards more sustainable and cost-effective manufacturing of high-purity pharmaceutical intermediates, ensuring a reliable supply chain for complex API precursors.
Patent CN113105402B discloses a groundbreaking preparation method for 3,4,5-trisubstituted 1,2,4-triazole compounds that addresses long-standing challenges in synthetic efficiency and environmental compliance. This novel approach leverages a non-metallic iodine-promoted strategy, eliminating the reliance on costly transition metal catalysts while maintaining high functional group tolerance. For R&D directors and procurement specialists, this technology represents a pivotal shift towards more sustainable and cost-effective manufacturing of high-purity pharmaceutical intermediates, ensuring a reliable supply chain for complex API precursors.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthetic routes for constructing polysubstituted 1,2,4-triazoles often suffer from significant operational complexities that hinder industrial adoption. Many established protocols rely heavily on transition metal catalysis, which introduces severe downstream processing burdens related to the removal of toxic metal residues to meet stringent regulatory limits for pharmaceutical ingredients. Furthermore, conventional methods frequently demand rigorous anhydrous and oxygen-free conditions, necessitating specialized equipment and inert atmosphere handling that drastically increases capital expenditure and operational overhead. The use of expensive reagents and the generation of substantial hazardous waste streams further exacerbate the economic and environmental footprint of these legacy processes. Consequently, scaling these reactions to commercial volumes often results in inconsistent yields and purity profiles, creating bottlenecks in the supply of critical drug intermediates.
The Novel Approach
In stark contrast, the methodology outlined in the patent utilizes a simple yet highly effective iodine-promoted cyclization strategy that operates under remarkably mild and practical conditions. By employing elemental iodine in conjunction with dimethyl sulfoxide (DMSO), the process facilitates a tandem oxidation-cyclization sequence that bypasses the need for sensitive metal catalysts entirely. 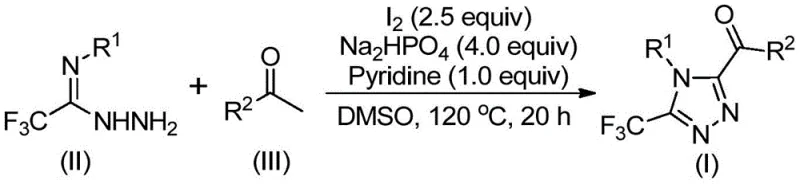 This innovation allows the reaction to proceed without strict exclusion of air or moisture, significantly simplifying reactor setup and operational protocols for plant engineers. The use of cheap and readily available starting materials, such as aryl ethyl ketones and trifluoroethylimide hydrazides, ensures that the cost of goods sold (COGS) remains competitive. Moreover, the reaction demonstrates excellent scalability, having been successfully expanded to gram levels with consistent performance, thereby offering a viable pathway for the commercial scale-up of complex pharmaceutical intermediates with reduced lead times.
This innovation allows the reaction to proceed without strict exclusion of air or moisture, significantly simplifying reactor setup and operational protocols for plant engineers. The use of cheap and readily available starting materials, such as aryl ethyl ketones and trifluoroethylimide hydrazides, ensures that the cost of goods sold (COGS) remains competitive. Moreover, the reaction demonstrates excellent scalability, having been successfully expanded to gram levels with consistent performance, thereby offering a viable pathway for the commercial scale-up of complex pharmaceutical intermediates with reduced lead times.
Mechanistic Insights into Iodine-Promoted Cyclization
The core of this synthetic breakthrough lies in the dual role of the iodine-DMSO system, which acts as both an oxidant and a promoter for the construction of the triazole ring. Mechanistically, the process initiates with the iodination and subsequent Kornblum oxidation of the aryl ethyl ketone substrate to generate a reactive aryl diketone intermediate in situ. This electrophilic species then undergoes a condensation reaction with the trifluoroethylimide hydrazide to form a hydrazone intermediate, setting the stage for the final ring closure. The presence of sodium dihydrogen phosphate and pyridine serves to buffer the reaction medium and facilitate the intramolecular cyclization step, driving the formation of the thermodynamically stable 1,2,4-triazole core. This cascade transformation is highly efficient, minimizing the formation of side products and ensuring a clean impurity profile that is crucial for downstream API synthesis.
From a structural perspective, the method exhibits remarkable versatility regarding the substitution patterns on the triazole ring, accommodating a wide array of electronic and steric environments. 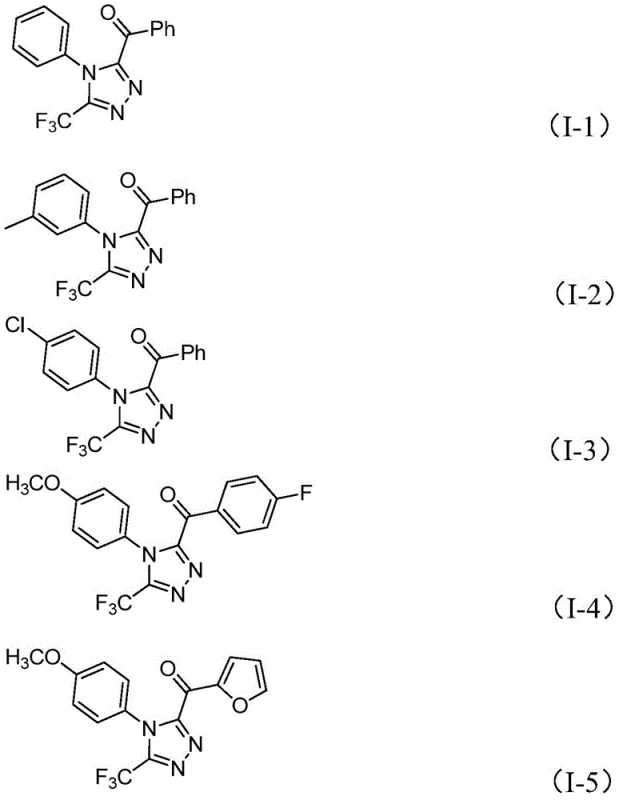 As demonstrated by the successful synthesis of derivatives I-1 through I-5, the protocol tolerates various substituents on both the N-aryl group (R1) and the acyl group (R2), including electron-donating methoxy groups, electron-withdrawing halogens, and bulky alkyl chains. This broad substrate scope is particularly valuable for medicinal chemists engaged in structure-activity relationship (SAR) studies, as it allows for the rapid generation of diverse compound libraries. The ability to introduce the trifluoromethyl group at the 3-position while simultaneously installing diverse acyl functionalities at the 5-position provides a powerful tool for optimizing the physicochemical properties of drug candidates without compromising synthetic feasibility.
As demonstrated by the successful synthesis of derivatives I-1 through I-5, the protocol tolerates various substituents on both the N-aryl group (R1) and the acyl group (R2), including electron-donating methoxy groups, electron-withdrawing halogens, and bulky alkyl chains. This broad substrate scope is particularly valuable for medicinal chemists engaged in structure-activity relationship (SAR) studies, as it allows for the rapid generation of diverse compound libraries. The ability to introduce the trifluoromethyl group at the 3-position while simultaneously installing diverse acyl functionalities at the 5-position provides a powerful tool for optimizing the physicochemical properties of drug candidates without compromising synthetic feasibility.
How to Synthesize 3,4,5-Trisubstituted 1,2,4-Triazole Efficiently
The operational simplicity of this protocol makes it an ideal candidate for technology transfer from laboratory to pilot plant scales. The procedure involves a straightforward two-stage heating process where reagents are added sequentially to a DMSO solution, eliminating the need for complex addition funnels or cryogenic cooling. The reaction progress can be easily monitored, and the workup involves standard filtration and silica gel chromatography techniques familiar to any process chemistry team. For detailed standard operating procedures and specific stoichiometric ratios optimized for maximum yield, please refer to the standardized synthesis guide below.
- Mix aryl ethyl ketone and iodine in DMSO, heating to 90-110°C for 4-6 hours to initiate oxidation.
- Add sodium dihydrogen phosphate, pyridine, and trifluoroethylimide hydrazide to the reaction mixture.
- Heat the solution to 110-130°C for 12-20 hours to complete the cyclization, followed by filtration and purification.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this iodine-promoted synthesis route offers tangible strategic advantages that directly impact the bottom line and operational resilience. The elimination of precious metal catalysts not only reduces raw material costs but also removes the necessity for expensive scavenging resins or complex extraction protocols required to lower metal content to ppm levels. This streamlining of the purification process translates into shorter batch cycle times and higher overall equipment effectiveness (OEE). Furthermore, the reliance on commodity chemicals like iodine and DMSO, which are globally sourced and abundant, mitigates the risk of supply chain disruptions associated with specialized reagents. The robustness of the reaction conditions ensures consistent quality output, reducing the rate of batch failures and enhancing the reliability of the supply chain for high-purity pharmaceutical intermediates.
- Cost Reduction in Manufacturing: The primary driver for cost optimization in this process is the complete avoidance of transition metal catalysts, which are often subject to volatile market pricing and require rigorous removal steps. By substituting these with inexpensive elemental iodine, the direct material costs are significantly lowered. Additionally, the simplified workup procedure reduces solvent consumption and labor hours associated with purification, leading to substantial operational savings. The high atom economy of the cyclization step further contributes to waste minimization, aligning with green chemistry principles that increasingly influence vendor selection criteria in the pharmaceutical sector.
- Enhanced Supply Chain Reliability: The starting materials for this synthesis, specifically aryl ethyl ketones and hydrazide derivatives, are widely available from multiple global suppliers, preventing single-source dependency risks. The reaction's tolerance to ambient conditions means that manufacturing does not require specialized inert atmosphere facilities, allowing for production across a broader range of contract manufacturing organizations (CMOs). This flexibility enhances supply security and allows for faster response times to fluctuating market demands. The proven scalability of the method ensures that volume requirements can be met without the need for extensive process re-engineering, securing long-term supply continuity.
- Scalability and Environmental Compliance: Scaling this process is inherently safer and more environmentally friendly due to the absence of pyrophoric reagents or high-pressure hydrogenation steps. The use of DMSO as a solvent, while requiring proper recovery systems, is well-established in industrial settings. The reduction in hazardous waste generation, particularly heavy metal sludge, simplifies waste disposal compliance and lowers environmental fees. This alignment with strict environmental, social, and governance (ESG) goals makes the resulting intermediates more attractive to multinational corporations committed to sustainable sourcing practices.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and beneficial effects reported in the patent documentation, providing clarity on process capabilities and limitations. Understanding these details is essential for evaluating the feasibility of integrating this route into existing manufacturing portfolios.
Q: Does this synthesis method require expensive transition metal catalysts?
A: No, the method described in patent CN113105402B utilizes elemental iodine as a promoter instead of toxic or expensive heavy metal catalysts, significantly simplifying downstream purification and reducing environmental impact.
Q: What are the optimal reaction conditions for this triazole formation?
A: The process operates effectively in dimethyl sulfoxide (DMSO) without strict anhydrous or oxygen-free conditions, typically requiring a two-stage heating profile between 90°C and 130°C over a total duration of approximately 20 hours.
Q: Is this method suitable for large-scale industrial production?
A: Yes, the protocol is designed for scalability, utilizing cheap and readily available starting materials like aryl ethyl ketones, and has been demonstrated to expand easily from gram-level laboratory synthesis to larger commercial batches.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3,4,5-Trisubstituted 1,2,4-Triazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient and scalable synthetic routes in the development of next-generation therapeutics. Our technical team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from benchtop discovery to industrial manufacturing is seamless. We are committed to delivering high-purity intermediates that meet stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation. Our expertise in heterocyclic chemistry allows us to optimize this iodine-promoted protocol further, tailoring it to specific client needs while maintaining the highest standards of quality and safety.
We invite potential partners to engage with our technical procurement team to discuss how this innovative synthesis method can be integrated into your supply chain. By requesting a Customized Cost-Saving Analysis, you can gain a deeper understanding of the economic benefits specific to your project volume. We encourage you to contact us for specific COA data and route feasibility assessments, ensuring that your project moves forward with a solid foundation of technical and commercial viability. Let us be your trusted partner in navigating the complexities of pharmaceutical intermediate manufacturing.