Advanced Synthesis of 2-(1,6,7,8-tetrahydro-2H-indeno-[5,4-b]furan-8-yl)acetonitrile for Ramelteon Production
The pharmaceutical industry continuously seeks robust synthetic pathways for Central Nervous System (CNS) therapeutics, particularly for insomnia treatments where safety and purity are paramount. Patent CN101824012B introduces a significant technological advancement in the synthesis of 2-(1,6,7,8-tetrahydro-2H-indeno-[5,4-b]furan-8-yl)acetonitrile, a pivotal chiral intermediate for the production of Ramelteon. Ramelteon, a tricyclic synthetic analog of melatonin, acts as a selective agonist for MT1 and MT2 receptors, offering a non-addictive solution for sleep onset insomnia without the side effects associated with traditional sedatives. The disclosed methodology addresses critical bottlenecks in existing manufacturing processes by replacing complex asymmetric hydrogenation steps with a more accessible amidation-dehydration sequence. This innovation not only streamlines the supply chain for this high-purity pharmaceutical intermediate but also aligns with modern green chemistry principles by reducing the reliance on precious metal catalysts and harsh reaction conditions.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of optically active Ramelteon has relied heavily on sophisticated and costly techniques such as homogeneous asymmetric catalytic hydrogenation using Ruthenium-BINAP complexes or enzymatic hydrolysis resolutions. These conventional routes present substantial challenges for commercial scale-up, primarily due to the exorbitant cost and limited availability of catalysts like [RuCl(benzene)(R)-BINAP]Cl and Ru(OAc)2-[(R)-BINAP]. Furthermore, these methods often necessitate absolute anhydrous solvent systems and high-pressure hydrogenation equipment, which increases capital expenditure and operational complexity. A major drawback is the potential for heavy metal contamination in the final Active Pharmaceutical Ingredient (API), requiring extensive and expensive purification steps to meet stringent regulatory limits. Additionally, the multi-step nature of these traditional pathways often results in lower overall yields and generates significant amounts of hazardous waste, complicating environmental compliance and driving up the total cost of goods sold.
The Novel Approach
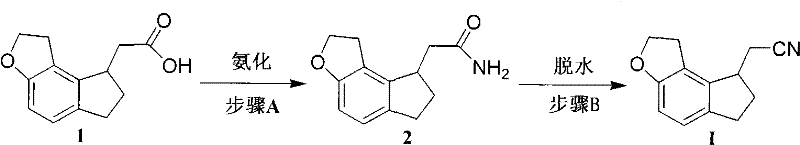
In stark contrast, the novel approach detailed in the patent utilizes a straightforward two-step transformation starting from 2-(1,6,7,8-tetrahydro-2H-indeno-[5,4-b]furan-8-yl)acetic acid or its optical isomers. The first step involves converting the carboxylic acid into an amide derivative via activation with acid halides, mixed anhydrides, or active esters, followed by ammonolysis. The second critical step is the dehydration of this amide to form the target nitrile intermediate using common dehydrating agents. This strategy effectively bypasses the need for expensive transition metal catalysts in the early stages. By employing reagents such as thionyl chloride, phosphorus oxychloride, or trifluoroacetic anhydride, the process achieves high conversion rates under mild thermal conditions ranging from -20°C to solvent reflux temperatures. This shift in synthetic logic drastically simplifies the operational workflow, reduces the risk of metal impurities, and enhances the overall economic viability of producing this key pharmaceutical building block.
Mechanistic Insights into Amide Dehydration and Coupling Chemistry
The core of this innovative synthesis lies in the efficient dehydration of the amide intermediate to the nitrile functionality. Mechanistically, this transformation is facilitated by strong dehydrating agents that activate the amide oxygen, rendering it a better leaving group. For instance, when using thionyl chloride (SOCl2), the oxygen of the amide attacks the sulfur atom, forming an imidoyl chloride intermediate which subsequently eliminates HCl to yield the nitrile. Alternatively, the use of trifluoroacetic anhydride (TFAA) activates the amide through the formation of an O-acyl imidate intermediate, which undergoes elimination to produce the cyano group. The choice of solvent plays a crucial role in stabilizing these intermediates; polar aprotic solvents like N,N-dimethylformamide (DMF) or tetrahydrofuran (THF) are often preferred to ensure homogeneity and optimal reaction kinetics. This mechanistic pathway is highly advantageous because it preserves the stereochemical integrity of the chiral center at the 8-position of the indeno-furan ring, ensuring that the optical purity of the starting material is carried through to the final nitrile product without racemization.
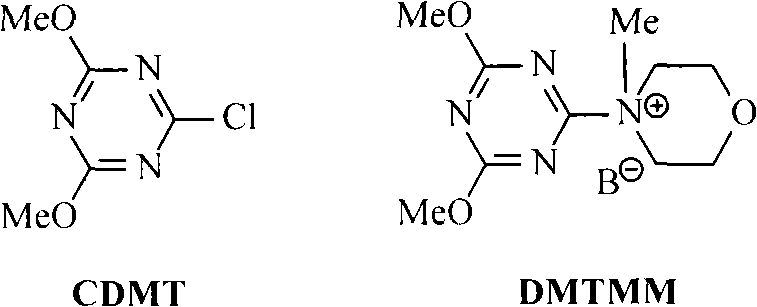
Furthermore, the downstream conversion of the nitrile to Ramelteon involves a reduction to the primary amine followed by acylation. The patent highlights the use of advanced coupling agents such as 2-chloro-4,6-dimethoxy-1,3,5-triazine (CDMT) and its morpholine salt derivative (DMTMM) for the final amide bond formation. These reagents operate via the formation of an active ester species in situ, which reacts rapidly with the amine nucleophile. This coupling strategy is superior to traditional carbodiimide methods as it minimizes the formation of urea byproducts and allows the reaction to proceed efficiently at near-neutral pH conditions. The use of DMTMM, in particular, offers enhanced solubility and reactivity, facilitating high-yield couplings even in complex molecular environments. This precise control over the acylation step ensures that the final API meets the rigorous purity specifications required for clinical applications, minimizing the presence of regioisomers or over-acylated impurities.
How to Synthesize 2-(1,6,7,8-tetrahydro-2H-indeno-[5,4-b]furan-8-yl)acetonitrile Efficiently
The synthesis of this critical intermediate is designed to be operationally simple yet chemically robust, making it ideal for transfer from laboratory scale to pilot plant operations. The process begins with the activation of the carboxylic acid precursor, which can be achieved using a variety of chlorinating agents or coupling reagents depending on the specific substrate sensitivity. Following the formation of the amide, the dehydration step is conducted under controlled thermal conditions to maximize yield while preventing thermal degradation of the sensitive indeno-furan scaffold. Detailed standard operating procedures regarding stoichiometry, temperature profiles, and workup protocols are essential for reproducibility. For a comprehensive guide on the specific experimental conditions and purification techniques validated in the patent examples, please refer to the standardized synthesis steps outlined below.
- Convert 2-(1,6,7,8-tetrahydro-2H-indeno-[5,4-b]furan-8-yl)acetic acid into the corresponding amide derivative using activation agents like thionyl chloride or mixed anhydrides followed by ammonolysis.
- Perform a dehydration reaction on the amide intermediate using dehydrating agents such as thionyl chloride, phosphorus oxychloride, or trifluoroacetic anhydride to yield the target acetonitrile compound.
- Optionally reduce the nitrile to the amine and couple with propionic acid using coupling agents like CDMT or DMTMM to finalize the Ramelteon API structure.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement and supply chain perspective, this patented methodology offers transformative benefits that directly impact the bottom line and operational resilience. By shifting away from precious metal catalysts and high-pressure infrastructure, manufacturers can significantly reduce capital investment and raw material costs. The reliance on commodity chemicals such as thionyl chloride and ammonia ensures a stable and predictable supply chain, mitigating the risks associated with sourcing specialized chiral catalysts that may have long lead times or geopolitical supply constraints. Moreover, the simplified purification processes reduce solvent consumption and waste generation, leading to substantial cost savings in waste disposal and environmental compliance. This efficiency translates into a more competitive pricing structure for the final API, allowing pharmaceutical companies to offer more affordable treatments to patients while maintaining healthy profit margins.
- Cost Reduction in Manufacturing: The elimination of expensive Ruthenium-based catalysts and the avoidance of high-pressure hydrogenation equipment result in drastic reductions in both fixed and variable manufacturing costs. The use of inexpensive dehydrating agents and standard solvents further lowers the cost of goods, while the high yields reported in the patent examples minimize material loss. Additionally, the simplified workup procedures reduce labor hours and energy consumption associated with distillation and chromatography, contributing to overall operational efficiency.
- Enhanced Supply Chain Reliability: Sourcing raw materials for this process is significantly more reliable compared to traditional methods, as the key reagents are bulk commodities available from multiple global suppliers. This diversification of the supply base reduces the risk of production stoppages due to single-source dependencies. The robustness of the reaction conditions also means that the process is less susceptible to variations in raw material quality, ensuring consistent output and reliable delivery schedules for downstream API manufacturers.
- Scalability and Environmental Compliance: The mild reaction conditions and absence of toxic heavy metals make this process inherently safer and easier to scale from kilogram to multi-ton production levels. The reduced generation of hazardous waste aligns with increasingly strict environmental regulations, lowering the burden of waste treatment and permitting. This 'green' profile not only enhances the corporate sustainability image but also future-proofs the manufacturing site against tightening environmental legislation, ensuring long-term operational continuity.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis route. These answers are derived directly from the experimental data and technical specifications provided in the patent documentation, offering clarity on purity, scalability, and regulatory compliance. Understanding these details is crucial for technical teams evaluating the feasibility of adopting this new methodology for commercial production.
Q: What are the advantages of this new synthesis route over traditional methods?
A: This method eliminates the need for expensive homogeneous asymmetric catalytic hydrogenation catalysts like Ruthenium-BINAP complexes. It operates under milder conditions, avoids heavy metal contamination, and significantly simplifies the post-treatment process, leading to higher chemical and optical purity.
Q: Which dehydrating agents are preferred for the nitrile formation step?
A: The patent highlights several effective dehydrating agents including thionyl chloride, phosphorus oxychloride, trifluoroacetic anhydride, and 2,4,6-trichloro-1,3,5-s-triazine. Thionyl chloride and trifluoroacetic anhydride are particularly noted for achieving high yields under reflux conditions.
Q: Is this process suitable for large-scale commercial production?
A: Yes, the process is designed for scalability. It utilizes readily available raw materials, avoids high-pressure hydrogenation in the initial steps, and employs standard solvents like chloroform, toluene, and tetrahydrofuran, making it highly adaptable for industrial manufacturing from 100 kgs to multi-ton scales.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-(1,6,7,8-tetrahydro-2H-indeno-[5,4-b]furan-8-yl)acetonitrile Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of high-quality intermediates in the development of life-saving CNS medications. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with precision and consistency. We adhere to stringent purity specifications and operate rigorous QC labs equipped with state-of-the-art analytical instrumentation to guarantee that every batch of 2-(1,6,7,8-tetrahydro-2H-indeno-[5,4-b]furan-8-yl)acetonitrile meets or exceeds global pharmacopoeial standards. Our commitment to quality assurance extends beyond mere compliance; we actively collaborate with our partners to optimize processes for maximum efficiency and minimal environmental impact.
We invite you to engage with our technical procurement team to discuss how this innovative synthesis route can benefit your specific project requirements. By requesting a Customized Cost-Saving Analysis, you can gain valuable insights into the potential economic advantages of switching to this metal-free methodology. We encourage you to contact us today to obtain specific COA data and route feasibility assessments tailored to your production goals, ensuring a seamless transition to a more sustainable and cost-effective supply chain for your Ramelteon manufacturing needs.