Advanced Manufacturing of Nirmatrelvir Intermediates via Active Anhydride Technology
Introduction to Next-Generation Antiviral Synthesis
The global pharmaceutical landscape has witnessed an unprecedented demand for effective antiviral therapeutics, specifically targeting SARS-CoV-2. At the heart of this therapeutic revolution lies Nirmatrelvir, the protease inhibitor component of the widely recognized Paxlovid regimen. However, the complex molecular architecture of Nirmatrelvir presents significant challenges in terms of synthetic efficiency and stereochemical control. Patent CN114989045A introduces a groundbreaking methodology that addresses these critical bottlenecks by designing novel key intermediates, designated as Intermediate I and Intermediate II. This technical disclosure outlines a robust preparation method that leverages the conversion of carboxylic acids into active anhydrides, facilitating a smoother, higher-yielding pathway to the final active pharmaceutical ingredient (API). By optimizing the coupling strategies and protecting group manipulations, this innovation offers a viable solution for manufacturers seeking to enhance their production capabilities.
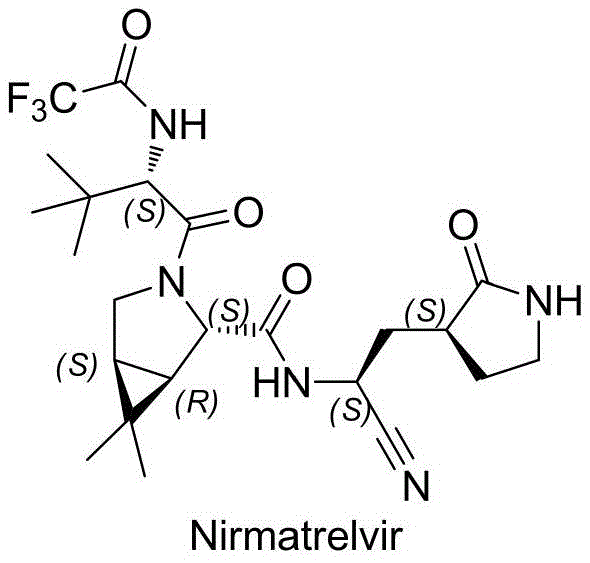
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art synthetic routes, such as those reported in foundational scientific literature like Science (2021), have demonstrated considerable limitations when subjected to rigorous industrial scrutiny. The original research indicates that repeating these established synthetic pathways often results in extremely low product yields and the generation of substantial quantities of difficult-to-remove byproducts. Furthermore, a pervasive issue in these conventional methods is the varying degrees of racemization risk, which compromises the optical purity of the final drug substance. In the context of regulatory compliance for a reliable pharmaceutical intermediate supplier, maintaining strict enantiomeric excess is non-negotiable. The reliance on harsh reaction conditions or less selective coupling reagents in traditional protocols frequently leads to epimerization at sensitive chiral centers, necessitating costly and yield-depleting purification steps that hinder commercial viability.
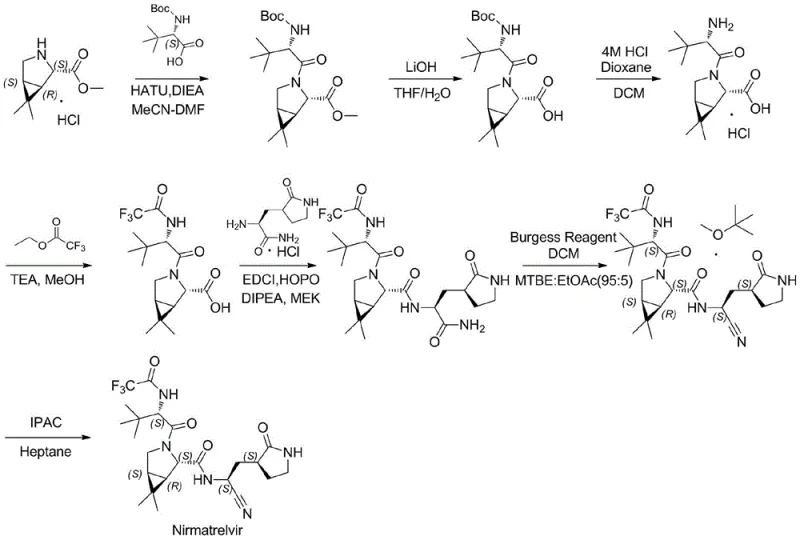
The Novel Approach
In stark contrast to the deficiencies of legacy methods, the novel approach detailed in the patent utilizes a strategic activation of carboxylic acids via mixed anhydride formation. By employing isobutyl chloroformate in the presence of N-methylmorpholine, the synthesis generates highly reactive yet controllable Intermediate I and Intermediate II. This methodology allows for coupling reactions to proceed under remarkably mild conditions, typically maintained between 0°C and 5°C. Such thermal control is pivotal for preserving the stereochemical integrity of the molecule. The result is a synthetic route that not only simplifies the operational workflow but also drastically improves the overall yield, reportedly achieving a total yield of over 62% compared to approximately 50% in prior art. This leap in efficiency translates directly to cost reduction in pharmaceutical intermediates manufacturing, making the process economically superior for large-scale operations.
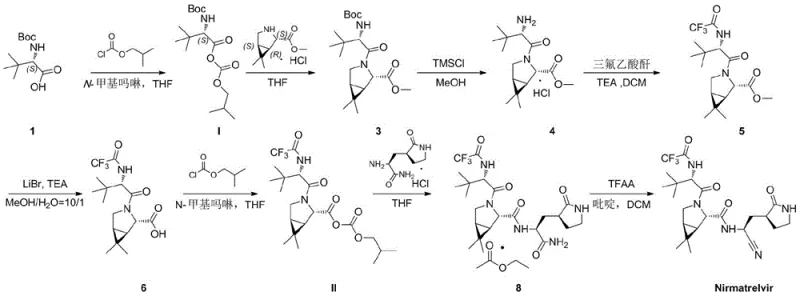
Mechanistic Insights into Mixed Anhydride Activation
The core mechanistic advantage of this technology lies in the in situ generation of mixed carbonic-carboxylic anhydrides. When the carboxylic acid precursor reacts with isobutyl chloroformate, it forms an activated species that is highly susceptible to nucleophilic attack by the amine component. Unlike carbodiimide-based couplings which can generate stable urea byproducts that are difficult to separate, the byproducts of this anhydride method (such as isobutyl carbonate derivatives) are generally more manageable or volatile. The reaction kinetics are optimized by the use of N-methylmorpholine as a base, which scavenges the generated HCl without promoting base-catalyzed racemization. This delicate balance ensures that the nucleophilic substitution occurs rapidly at the carbonyl carbon of the anhydride before any significant epimerization of the alpha-carbon can take place. Consequently, the high-purity pharmaceutical intermediate obtained retains the critical (S) and (R) configurations required for biological activity.
Furthermore, the downstream functional group transformations are meticulously designed to support this high-fidelity activation strategy. For instance, the hydrolysis of the methyl ester to the free acid (Compound 6) utilizes lithium bromide and triethylamine in a methanol-water system. This specific reagent choice avoids the use of strong inorganic bases like sodium hydroxide, which could otherwise hydrolyze the sensitive trifluoroacetamide group or induce lactam ring opening. The subsequent re-activation of this acid to form Intermediate II follows the same anhydride logic, ensuring consistency throughout the synthesis. This mechanistic coherence minimizes the formation of impurities related to the synthetic route, thereby simplifying the purification profile and enhancing the overall robustness of the commercial scale-up of complex pharmaceutical intermediates.
How to Synthesize Nirmatrelvir Efficiently
The synthesis of Nirmatrelvir via this patented route involves a sequence of precise activation, coupling, and cyclization steps that prioritize yield and purity. The process begins with the activation of a Boc-protected amino acid, followed by coupling with a bicyclic proline derivative. Subsequent steps involve careful deprotection and re-protection to install the trifluoroacetamide moiety, followed by ester hydrolysis and a second activation cycle to attach the nitrile-containing warhead. The final step utilizes trifluoroacetic anhydride to effect a dehydration cyclization, yielding the target API. For process chemists looking to implement this technology, the detailed standardized synthesis steps are outlined below to ensure reproducibility and safety.
- Activate Boc-protected amino acid with isobutyl chloroformate and N-methylmorpholine to form Intermediate I, then couple with the bicyclic amine ester.
- Perform sequential deprotection using TMSCl/MeOH and acylation with trifluoroacetic anhydride to install the trifluoroacetamide group, followed by ester hydrolysis.
- Convert the resulting acid to Intermediate II using isobutyl chloroformate, couple with the nitrile-containing amine, and finalize with TFAA-mediated cyclization.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this synthetic route offers tangible strategic benefits beyond mere technical elegance. The primary advantage is the significant optimization of the cost structure associated with API production. By eliminating the need for expensive transition metal catalysts or exotic coupling reagents that require extensive removal protocols, the process inherently lowers the cost of goods sold (COGS). The high yields reported at each step mean that less raw material is wasted, and the throughput per batch is maximized. This efficiency is critical for reducing lead time for high-purity pharmaceutical intermediates, allowing manufacturers to respond more agilely to market demands without the burden of excessive inventory buffers caused by low-yielding processes.
- Cost Reduction in Manufacturing: The utilization of commodity chemicals such as isobutyl chloroformate and N-methylmorpholine replaces costlier activation agents. Furthermore, the mild reaction conditions reduce energy consumption for heating or extreme cooling, while the simplified purification requirements lower solvent usage and waste disposal costs. These factors combine to create a leaner manufacturing process that delivers substantial economic value without compromising quality standards.
- Enhanced Supply Chain Reliability: The raw materials required for this synthesis are widely available in the global chemical market, reducing the risk of supply disruptions associated with specialized reagents. The robustness of the reaction conditions means that the process is less sensitive to minor fluctuations in operating parameters, ensuring consistent batch-to-batch quality. This reliability is essential for maintaining a continuous supply of critical antiviral medications to the global healthcare system.
- Scalability and Environmental Compliance: The absence of heavy metals and the use of relatively benign solvents align with modern green chemistry principles. The process generates fewer hazardous byproducts, simplifying wastewater treatment and environmental compliance. Additionally, the straightforward nature of the unit operations facilitates seamless scaling from pilot plant to multi-ton commercial production, ensuring that supply can meet surging global demand efficiently.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this advanced synthetic pathway. These insights are derived directly from the patent specifications and are intended to clarify the operational benefits for potential manufacturing partners. Understanding these nuances is key to evaluating the feasibility of integrating this technology into existing production lines.
Q: How does the active anhydride method improve yield compared to conventional routes?
A: By converting carboxylic acids into active mixed anhydrides using isobutyl chloroformate, the reaction proceeds under milder conditions (0-5°C), significantly reducing byproduct formation and increasing overall yield compared to harsher activation methods.
Q: Does this synthesis route minimize the risk of racemization?
A: Yes, the use of mild temperatures and specific activation reagents like isobutyl chloroformate preserves the stereochemical integrity of the chiral centers, effectively mitigating the risk of racemization often seen in high-temperature peptide couplings.
Q: Is this process suitable for large-scale commercial production?
A: Absolutely. The process utilizes commercially available raw materials and avoids complex transition metal catalysts, making it highly scalable, cost-effective, and easier to purify for industrial manufacturing of pharmaceutical intermediates.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Nirmatrelvir Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient and scalable synthesis routes for life-saving antiviral medications. Our team of expert process chemists has thoroughly analyzed the technological potential of the active anhydride method described in Patent CN114989045A. As a leading CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our state-of-the-art facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications, ensuring that every batch of Nirmatrelvir intermediate we produce adheres to the highest international regulatory standards. We are committed to translating innovative patent technologies into reliable commercial realities.
We invite pharmaceutical companies and contract manufacturers to collaborate with us to optimize their supply chains. By leveraging our expertise in peptide coupling and heterocyclic chemistry, we can help you achieve significant process improvements. Please contact our technical procurement team to request a Customized Cost-Saving Analysis. We are ready to provide specific COA data and comprehensive route feasibility assessments to demonstrate how our manufacturing capabilities can support your Nirmatrelvir production goals effectively.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →