Advanced Synthesis of Trilaciclib Intermediates: A Cost-Effective Route for Commercial Scale-Up
Advanced Synthesis of Trilaciclib Intermediates: A Cost-Effective Route for Commercial Scale-Up
The pharmaceutical industry is constantly seeking more efficient pathways for the production of complex oncology therapeutics, and the synthesis of Trilaciclib (also known as Trasipride or G1T28) represents a significant area of focus due to its role as a CDK4/6 inhibitor. Patent CN115477653A, published in late 2022, introduces a groundbreaking preparation method for a key intermediate of Trilaciclib and the final active pharmaceutical ingredient itself. This technology addresses critical bottlenecks in existing manufacturing processes by utilizing 2-amino-4-chloropyrimidine-5-carbaldehyde as a starting material and employing a strategic sequence of protection, substitution, and cyclization reactions. The structural complexity of Trilaciclib, featuring a spiro-piperidine core fused with a pyrrolo-pyrimidinone system, demands high precision in synthetic design to ensure purity and yield. 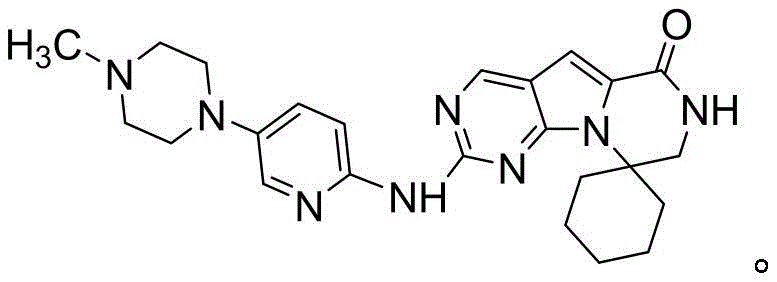
This new methodology stands out by drastically simplifying the synthetic landscape, moving away from the cumbersome multi-step protections and deprotections that characterized earlier generations of synthesis. By leveraging robust base-catalyzed transformations and avoiding hazardous reagents, this patent offers a pathway that is not only chemically elegant but also commercially viable for large-scale operations. For procurement and supply chain leaders, the implications are profound, as the shift towards simpler, safer chemistry often correlates with reduced operational risks and lower overall production costs. The ability to produce high-purity intermediates through a controlled, four-step sequence positions this technology as a superior alternative for reliable pharmaceutical intermediate supplier networks aiming to support the growing demand for myeloprotective agents in chemotherapy.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior to the innovations disclosed in CN115477653A, the synthesis of Trilaciclib was plagued by significant inefficiencies that hindered cost-effective manufacturing. The original routes, such as those published by G1 Therapeutics or detailed in patents like CN 109789142B, relied heavily on frequent protection and deprotection of functional groups, leading to an excessive number of reaction steps. 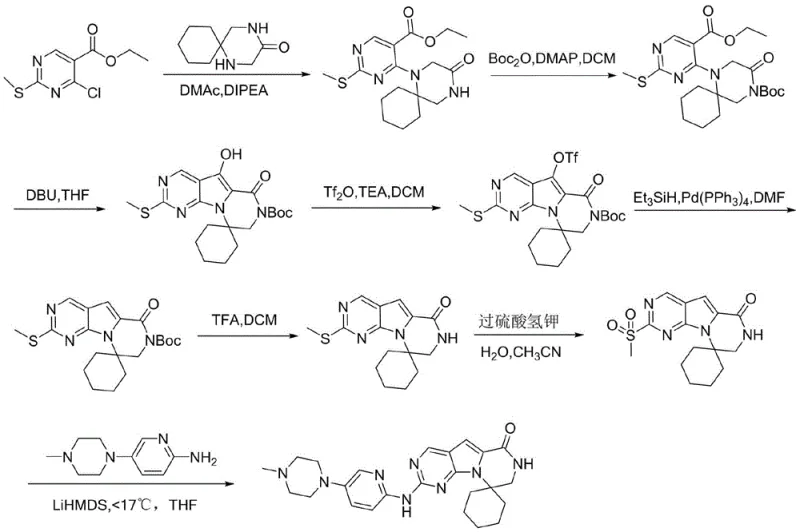
Furthermore, these conventional methods invariably depended on the use of expensive palladium catalysts for key coupling reactions, which not only inflated raw material costs but also introduced challenges related to heavy metal residue removal—a critical quality attribute for API manufacturing. Another prevalent route, disclosed in patents such as CN114014864A, attempted to reduce step counts but still suffered from the inevitable use of costly palladium systems and generated difficult-to-remove byproducts during intermediate stages. These impurities severely impacted the final yield and purity, necessitating complex purification protocols that further eroded profit margins and extended lead times for high-purity pharmaceutical intermediates.
The Novel Approach
In stark contrast to the convoluted legacy processes, the novel approach outlined in the present patent streamlines the entire synthesis into just four distinct chemical reactions. The strategy initiates with the selective protection of the amino group on the pyrimidine aldehyde using di-tert-butyl dicarbonate ((Boc)2O) under mild alkaline conditions, setting the stage for a highly selective substitution. 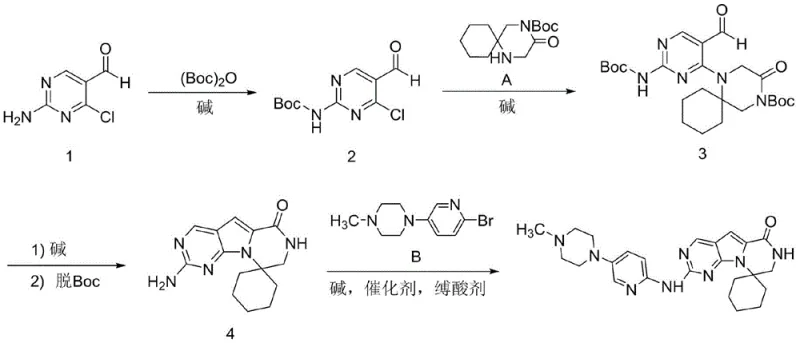
Subsequent steps involve a direct substitution with a spiro-piperidine derivative, followed by a clever intramolecular cyclization that simultaneously constructs the core ring system and removes protecting groups upon pH adjustment. This telescoping of cyclization and deprotection is a masterstroke of process chemistry, eliminating isolated intermediate handling and reducing solvent waste. The final step employs a nucleophilic substitution to attach the pyridine-piperazine side chain, crucially utilizing copper catalysis instead of palladium. This shift not only reduces cost reduction in API manufacturing but also simplifies the impurity profile, ensuring that the final product meets stringent regulatory standards with greater ease and consistency.
Mechanistic Insights into Base-Catalyzed Cyclization and Copper-Mediated Coupling
The heart of this synthetic innovation lies in the mechanistic efficiency of the cyclization and final coupling steps, which drive the overall yield and purity of the process. In Step S3, the intramolecular cyclization is triggered by a strong base, such as potassium tert-butoxide or sodium hydride, which deprotonates the active methylene position adjacent to the carbonyl group. This generates a nucleophilic enolate species that attacks the electrophilic center on the pyrimidine ring, closing the pyrrole ring to form the fused tricyclic core. The reaction is conducted in polar aprotic solvents like THF or acetonitrile at room temperature, allowing for precise control over the reaction kinetics and minimizing thermal degradation of sensitive intermediates.
Following cyclization, the process ingeniously integrates the removal of the Boc protecting groups by simply adjusting the pH to an acidic range (pH 2-3) during the workup. This dual-function step prevents the accumulation of protected byproducts and ensures that the amine functionality is exposed and ready for the final coupling without requiring a separate deprotection reactor run. The final Step S4 utilizes a copper-catalyzed nucleophilic aromatic substitution (or related coupling mechanism depending on the specific leaving group activation) to attach the 5-(4-methylpiperazin-1-yl)pyridin-2-amine moiety. The use of copper iodide or cuprous iodide, combined with bases like potassium tert-butoxide and ligands such as N,N'-dibenzyl oxalyl diamine, facilitates this bond formation under relatively mild heating (60 to 110°C). This mechanistic pathway avoids the harsh conditions often associated with palladium cross-couplings, thereby preserving the integrity of the spiro-cycle and preventing ring-opening side reactions.
How to Synthesize Trilaciclib Efficiently
The synthesis of Trilaciclib via this patented route offers a robust framework for process chemists aiming to establish a scalable manufacturing line. The procedure begins with the protection of 2-amino-4-chloropyrimidine-5-carbaldehyde, followed by coupling with the spiro-piperidine ketone, cyclization, and final amination. Each step has been optimized for high conversion and ease of isolation, typically involving crystallization or simple extraction workups. For R&D teams looking to implement this technology, the detailed standardized synthesis steps are provided below to ensure reproducibility and compliance with the patent's preferred embodiments.
- Protect the amino group of 2-amino-4-chloropyrimidine-5-carbaldehyde using (Boc)2O and a base like potassium carbonate in THF at 60°C.
- Perform a substitution reaction between the protected aldehyde and a spiro-piperidine derivative under alkaline conditions to form the coupled intermediate.
- Execute intramolecular cyclization using a strong base such as potassium tert-butoxide, followed by acidification to remove protecting groups.
- Conduct the final nucleophilic substitution with a pyridine-piperazine derivative using a copper catalyst and base to yield Trilaciclib.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this synthesis route offers transformative benefits for procurement managers and supply chain heads tasked with optimizing the sourcing of oncology intermediates. The most immediate impact is seen in the drastic simplification of the supply chain for raw materials; by eliminating the dependency on precious palladium catalysts, manufacturers can insulate themselves from the volatile pricing and geopolitical supply risks associated with platinum group metals. Instead, the process relies on abundant and inexpensive base metals like copper and common organic bases, which are readily available from multiple global suppliers, ensuring enhanced supply chain reliability and continuity of supply even during market disruptions.
- Cost Reduction in Manufacturing: The economic advantages of this route are driven primarily by the reduction in step count and the elimination of expensive catalytic systems. By condensing the synthesis into four high-yielding steps, the cumulative loss of material inherent in multi-step processes is significantly minimized, leading to a higher overall throughput of the final API. Furthermore, the avoidance of palladium removes the need for costly scavenging resins and extensive analytical testing for heavy metal residues, which are mandatory and expensive parts of the release criteria for injectable drugs. The use of simple workup procedures, such as acid-base extractions and crystallizations, reduces solvent consumption and energy usage compared to complex chromatographic purifications required by older methods.
- Enhanced Supply Chain Reliability: The reliance on commodity chemicals such as potassium carbonate, triethylamine, and THF ensures that the production of Trilaciclib intermediates is not bottlenecked by specialized reagent availability. This democratization of raw materials allows for flexible sourcing strategies, where procurement teams can leverage competitive bidding among multiple vendors for standard solvents and bases. Additionally, the robustness of the reaction conditions—operating at moderate temperatures and pressures—reduces the risk of batch failures due to equipment limitations or operator error, thereby stabilizing production schedules and reducing lead time for high-purity pharmaceutical intermediates.
- Scalability and Environmental Compliance: The green chemistry attributes of this process align perfectly with modern environmental, social, and governance (ESG) goals. The reduction in reaction steps directly correlates to a lower E-factor (mass of waste per mass of product), as fewer solvents and reagents are consumed per kilogram of output. The absence of toxic heavy metals simplifies waste stream treatment, lowering the cost of effluent disposal and reducing the environmental footprint of the manufacturing facility. This scalability makes the process ideal for commercial scale-up of complex pharmaceutical intermediates, allowing manufacturers to ramp up from pilot plant quantities to multi-ton annual production with minimal process re-engineering.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and claims within patent CN115477653A, providing a clear picture of the operational realities and advantages of this novel route for stakeholders evaluating its adoption.
Q: How does this new synthesis route reduce costs compared to prior art?
A: The novel route described in patent CN115477653A eliminates the need for expensive palladium catalysts used in previous methods, replacing them with more economical copper catalysts and common bases. Additionally, the reduction in synthetic steps from multiple tedious protections to a streamlined 4-step process significantly lowers raw material consumption and processing time.
Q: What are the critical reaction conditions for the cyclization step?
A: The intramolecular cyclization (Step S3) requires a strong base such as potassium tert-butoxide or sodium hydride in solvents like THF or acetonitrile. The reaction is typically conducted at room temperature for 6 to 10 hours, followed by careful pH adjustment to 2-3 to remove protecting groups effectively.
Q: Is this process suitable for large-scale industrial production?
A: Yes, the process is designed for industrial scalability. It utilizes readily available raw materials, avoids dangerous reagents, and features uniform, controllable reaction conditions. The workup procedures involve standard crystallization and extraction techniques, facilitating easy scale-up from laboratory to commercial tonnage.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Trilaciclib Supplier
At NINGBO INNO PHARMCHEM, we recognize the strategic importance of efficient synthetic routes in the competitive landscape of oncology drug development. Our team of expert process chemists has thoroughly analyzed the technology disclosed in CN115477653A and possesses the extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production required to bring this chemistry to life. We are committed to delivering high-purity Trilaciclib intermediates that meet stringent purity specifications, utilizing our rigorous QC labs to ensure every batch complies with the highest international standards for safety and efficacy.
We invite pharmaceutical partners to collaborate with us to leverage this cost-effective synthesis technology for their supply chains. By engaging with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage you to contact us today to obtain specific COA data and route feasibility assessments, ensuring that your project benefits from the most advanced and economically viable manufacturing solutions available in the market.