Scalable Iron-Catalyzed Synthesis of 2-Trifluoromethyl Quinazolinones for Advanced Pharmaceutical Manufacturing
Introduction to Advanced Quinazolinone Synthesis
The development of efficient synthetic routes for nitrogen-containing heterocycles remains a cornerstone of modern medicinal chemistry, particularly for scaffolds exhibiting potent biological activities. Patent CN111675662B discloses a groundbreaking preparation method for 2-trifluoromethyl substituted quinazolinone compounds, a class of molecules renowned for their anti-cancer, anticonvulsant, and anti-inflammatory properties. The introduction of the trifluoromethyl group into these heterocyclic systems significantly enhances metabolic stability, lipophilicity, and bioavailability, making them highly desirable candidates for drug discovery programs. This patent presents a novel approach that overcomes the limitations of traditional methods by utilizing readily available starting materials and an inexpensive iron catalyst, thereby addressing critical needs for cost reduction in pharmaceutical intermediate manufacturing.
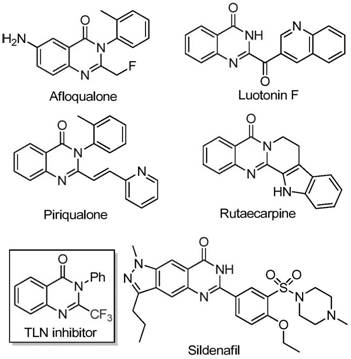
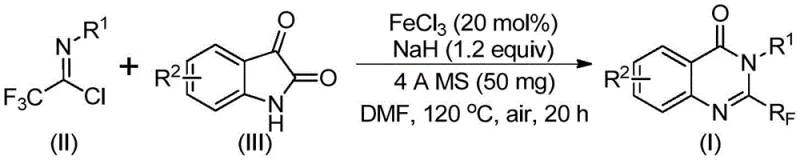
Historically, the synthesis of trifluoromethyl-substituted quinazolinones has relied on cyclization reactions involving expensive synthons such as trifluoroacetic anhydride or ethyl trifluoroacetate with substrates like anthranilamide. These conventional pathways are often plagued by severe reaction conditions, narrow substrate scope, and moderate yields, creating bottlenecks for reliable pharmaceutical intermediate suppliers. The disclosed invention shifts this paradigm by employing trifluoroethylimidoyl chloride and isatin derivatives as key building blocks. This strategic selection of precursors not only lowers the raw material costs but also streamlines the synthetic sequence, offering a robust platform for the commercial scale-up of complex pharmaceutical additives and active ingredients.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthetic methodologies for constructing the quinazolinone core with a trifluoromethyl moiety frequently encounter substantial hurdles that impede their utility in large-scale production. Prior art methods typically necessitate the use of harsh reagents and extreme temperatures, which can lead to the decomposition of sensitive functional groups and the formation of complex impurity profiles. Furthermore, the reliance on precious metal catalysts or stoichiometric amounts of expensive fluorinating agents drives up the overall production cost, making the final API economically unviable for certain therapeutic areas. The narrow tolerance for substituents on the aromatic ring often requires multi-step protection and deprotection sequences, drastically increasing the lead time for high-purity pharmaceutical intermediates and complicating the supply chain logistics.
The Novel Approach
In stark contrast, the method described in CN111675662B utilizes a synergistic combination of ferric chloride and sodium hydride to facilitate a cascade cyclization reaction under relatively mild conditions. By leveraging the unique reactivity of trifluoroethylimidoyl chloride, the process enables the direct construction of the quinazolinone skeleton from isatin derivatives with high atom economy. This novel approach eliminates the need for pre-functionalized anthranilic acid derivatives, thereby shortening the synthetic route and reducing waste generation. The ability to conduct the reaction in common polar aprotic solvents like DMF, coupled with the use of earth-abundant iron, represents a significant leap forward in sustainable chemistry, aligning perfectly with the industry's push towards greener manufacturing processes.
Mechanistic Insights into FeCl3-Catalyzed Cyclization
The mechanistic pathway of this transformation involves a sophisticated interplay between base-promoted nucleophilic substitution and transition metal-catalyzed decarbonylation. Initially, sodium hydride acts as a strong base to deprotonate the isatin nitrogen, generating a nucleophilic species that attacks the electrophilic carbon of the trifluoroethylimidoyl chloride. This step forms a key trifluoroacetamidine intermediate, setting the stage for the subsequent ring closure. The presence of 4A molecular sieves is crucial at this stage to sequester any trace moisture that could quench the reactive hydride or hydrolyze the imidoyl chloride, ensuring high conversion rates and consistent batch-to-batch reproducibility.
Following the initial coupling, the ferric chloride catalyst orchestrates an intramolecular cyclization accompanied by a decarbonylation event. The iron center likely coordinates with the carbonyl oxygen and the imine nitrogen, facilitating the extrusion of carbon monoxide and the aromatization of the pyrimidine ring to yield the final 2-trifluoromethyl quinazolinone product. This mechanism explains the remarkable functional group tolerance observed, as the iron catalyst operates through a specific coordination sphere that avoids non-specific radical pathways often associated with other transition metals. Understanding this mechanistic nuance is vital for R&D directors aiming to optimize the process further or adapt it for analogous heterocyclic systems.
How to Synthesize 2-Trifluoromethyl Quinazolinones Efficiently
The operational simplicity of this protocol makes it highly attractive for both laboratory-scale optimization and pilot plant operations. The procedure involves a straightforward one-pot sequence where reagents are combined in a specific order to maximize safety and yield. Critical parameters such as the temperature ramp from 40°C to 120°C and the reaction duration are optimized to balance the kinetics of the initial substitution with the thermodynamics of the cyclization step. For detailed standard operating procedures regarding reagent grades, addition rates, and specific workup protocols, please refer to the standardized synthesis guide below.
- Combine ferric chloride (20 mol%), sodium hydride (1.2 equiv), 4A molecular sieves, trifluoroethylimidoyl chloride, and isatin derivative in anhydrous DMF solvent.
- Stir the reaction mixture at 40°C for approximately 10 hours to initiate the nucleophilic attack and intermediate formation.
- Raise the temperature to 120°C and continue stirring under air for 18-20 hours to complete the decarbonylation and cyclization, followed by silica gel purification.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement perspective, this patented methodology offers compelling value propositions that directly impact the bottom line of pharmaceutical manufacturing projects. The shift from precious metal catalysts to inexpensive ferric chloride results in a drastic reduction in catalyst costs, while the use of commercially available isatins and imidoyl chlorides ensures a stable and diversified supply base. This reduces the risk of supply chain disruptions caused by the scarcity of specialized reagents, providing procurement managers with greater flexibility in vendor selection and negotiation. Additionally, the simplified purification process, which relies on standard column chromatography or crystallization rather than complex metal scavenging steps, significantly lowers downstream processing costs.
- Cost Reduction in Manufacturing: The elimination of expensive palladium or rhodium catalysts, combined with the use of commodity chemicals like DMF and sodium hydride, leads to substantial cost savings in raw material expenditure. The high yields reported across a wide range of substrates minimize material loss, further enhancing the overall process economics and allowing for more competitive pricing of the final active pharmaceutical ingredients.
- Enhanced Supply Chain Reliability: Since the key starting materials such as isatin and various aromatic amines are produced on a multi-ton scale globally, the dependency on niche suppliers is effectively removed. This abundance ensures consistent availability and shorter lead times, enabling supply chain heads to maintain leaner inventory levels while mitigating the risk of production delays due to raw material shortages.
- Scalability and Environmental Compliance: The reaction conditions are amenable to scale-up, having been demonstrated from gram to multi-gram levels without loss of efficiency. The use of iron, a non-toxic metal, simplifies waste disposal and regulatory compliance regarding heavy metal residues in the final drug product, aligning with stringent environmental standards and reducing the burden on waste treatment facilities.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation of this synthesis route in an industrial setting. These insights are derived directly from the experimental data and scope limitations outlined in the patent documentation, providing clarity on process robustness and applicability.
Q: What are the primary advantages of using FeCl3 over precious metal catalysts for quinazolinone synthesis?
A: The use of ferric chloride (FeCl3) offers significant economic advantages due to its low cost and earth-abundant nature compared to palladium or rhodium catalysts. Furthermore, iron catalysis often exhibits excellent functional group tolerance, reducing the need for extensive protecting group strategies and simplifying the overall synthetic route for complex pharmaceutical intermediates.
Q: Can this synthesis method accommodate diverse substituents on the isatin ring?
A: Yes, the patented methodology demonstrates broad substrate scope, successfully tolerating various substituents such as methyl, fluoro, bromo, and methoxy groups at different positions on the aromatic ring. This versatility allows for the rapid generation of diverse libraries of 2-trifluoromethyl quinazolinones for SAR studies without requiring major protocol adjustments.
Q: Is the reaction sensitive to moisture or air during the cyclization step?
A: The process utilizes 4A molecular sieves to maintain anhydrous conditions critical for the initial deprotonation steps. However, the subsequent high-temperature cyclization step is conducted under air, indicating that the iron-catalyzed oxidative decarbonylation is robust and does not require stringent inert atmosphere techniques, which greatly facilitates industrial scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Quinazolinone Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of adopting innovative synthetic technologies to stay ahead in the competitive pharmaceutical landscape. Our team of expert chemists has thoroughly evaluated the FeCl3-catalyzed route described in CN111675662B and possesses the extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. We are committed to delivering high-purity 2-trifluoromethyl quinazolinones that meet stringent purity specifications, supported by our rigorous QC labs and state-of-the-art analytical capabilities to ensure every batch exceeds client expectations.
We invite potential partners to engage with our technical procurement team to discuss how this cost-effective technology can be integrated into your specific drug development pipeline. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into the economic benefits of switching to this iron-catalyzed process. Contact us today to obtain specific COA data and comprehensive route feasibility assessments tailored to your project requirements, ensuring a seamless transition from bench-scale discovery to full-scale commercial manufacturing.