Advanced Palladium-Catalyzed Synthesis of 2-Trifluoromethyl Quinazolinone Derivatives for Pharmaceutical Applications
Advanced Palladium-Catalyzed Synthesis of 2-Trifluoromethyl Quinazolinone Derivatives for Pharmaceutical Applications
The development of efficient synthetic routes for heterocyclic compounds remains a cornerstone of modern medicinal chemistry, particularly for scaffolds exhibiting broad biological activity. Patent CN112125856A discloses a novel preparation method for 2-trifluoromethyl substituted quinazolinone derivatives, addressing critical challenges in current synthetic methodologies. Quinazolinone derivatives are privileged structures found in numerous natural products and pharmaceutical agents, possessing a spectrum of activities including anti-inflammatory, antiviral, antifungal, anticonvulsant, and anticancer properties. As illustrated in the structural diversity of known bioactive molecules, the incorporation of specific functional groups can drastically alter pharmacokinetic profiles. This patent introduces a transition metal palladium-catalyzed carbonylation tandem reaction that utilizes cheap and readily available starting materials, specifically o-iodoaniline and trifluoroacetimidoyl chloride, to construct this valuable heterocyclic core with high efficiency and operational simplicity.
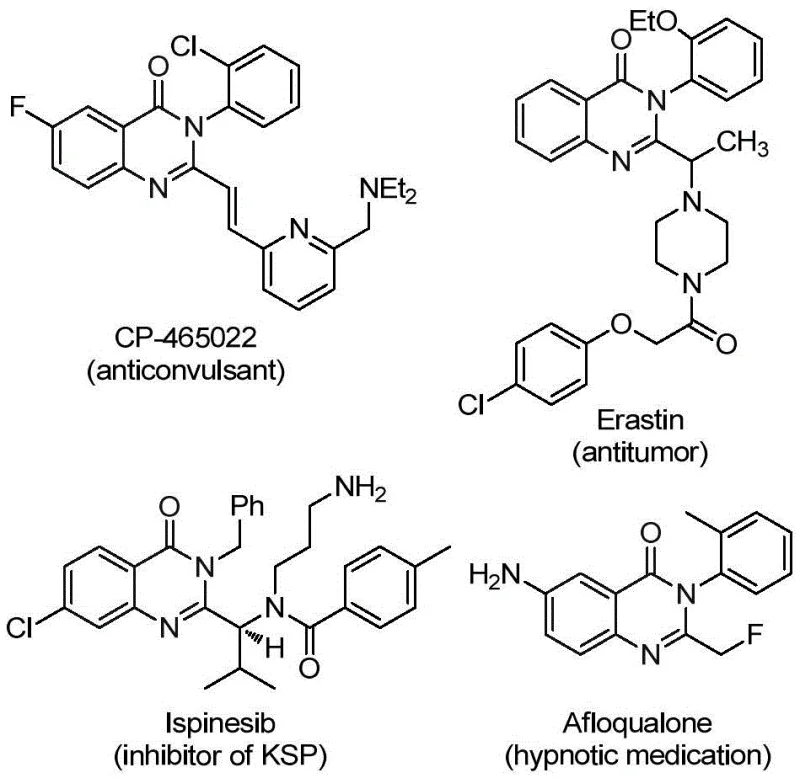
The significance of this technology lies not only in its chemical elegance but also in its potential to streamline the supply chain for complex pharmaceutical intermediates. By leveraging a solid carbon monoxide substitute, the process mitigates significant safety hazards associated with traditional gas-phase carbonylation, making it highly attractive for industrial scale-up. For R&D directors and procurement managers seeking reliable sources of high-purity intermediates, this methodology represents a substantial advancement in process chemistry, offering a robust pathway to access diverse libraries of fluorinated heterocycles essential for next-generation drug discovery programs.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of 2-trifluoromethyl substituted quinazolinone derivatives has been fraught with significant technical and logistical hurdles that impede large-scale production. Conventional literature reports typically rely on cyclization reactions involving anthranilamide with ethyl trifluoroacetate, trifluoroacetic anhydride, or trifluoroacetic acid under varying conditions. Alternative strategies include the cyclization of anthranilic acid esters with unstable trifluoroacetamides or the reaction of isatoic anhydride with trifluoroacetic anhydride. These traditional approaches suffer from several inherent disadvantages that limit their utility in a commercial setting. Firstly, the reaction conditions are often harsh, requiring extreme temperatures or pressures that can degrade sensitive functional groups. Secondly, many of the requisite starting materials are expensive or require complex pre-activation steps, driving up the overall cost of goods. Furthermore, these methods frequently exhibit low yields and narrow substrate scopes, meaning they fail to accommodate a wide variety of substituents, thereby restricting the chemical space available for medicinal chemists to explore during lead optimization phases.
The Novel Approach
In stark contrast to these legacy methods, the invention detailed in CN112125856A presents a transformative approach utilizing a palladium-catalyzed carbonylation tandem reaction. This novel strategy employs 1,3,5-tricarboxylate phenol ester (TFBen) as a solid carbon monoxide surrogate, effectively bypassing the need for toxic carbon monoxide gas while maintaining high reaction efficiency. The process operates under relatively mild conditions, typically at 90°C in an organic solvent such as tetrahydrofuran, using a catalytic system comprising Pd(PPh3)2Cl2 and dppp ligand. This methodology is exceptionally versatile, demonstrating excellent compatibility with various substituents on both the aromatic ring and the nitrogen atom. The ability to synthesize derivatives with different groups through simple substrate design significantly widens the practicability of the method. By avoiding the use of hazardous gases and expensive pre-activated substrates, this route offers a safer, more cost-effective, and scalable solution for producing high-purity 2-trifluoromethyl quinazolinones, directly addressing the pain points of conventional synthesis.
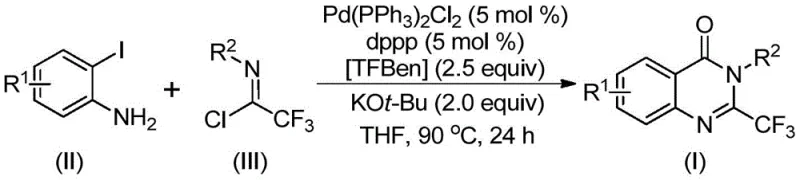
Mechanistic Insights into Palladium-Catalyzed Carbonylation Tandem Reaction
The mechanistic pathway of this transformation involves a sophisticated sequence of organometallic steps that ensure high regioselectivity and yield. The reaction likely initiates with a base-promoted intermolecular carbon-nitrogen bond coupling between the o-iodoaniline and the trifluoroacetimidoyl chloride, facilitated by potassium tert-butoxide, to generate a trifluoroacetamidine derivative intermediate. Subsequently, the palladium catalyst undergoes oxidative addition into the carbon-iodine bond of the aromatic ring, forming a divalent palladium species. Under the heating conditions employed, the solid CO surrogate TFBen decomposes to release carbon monoxide in situ. This generated carbon monoxide then inserts into the carbon-palladium bond to form a crucial acyl-palladium intermediate. The presence of the base further promotes the formation of a palladium-nitrogen bond, leading to the construction of a seven-membered ring palladium intermediate. Finally, a reductive elimination step occurs, releasing the final 2-trifluoromethyl substituted quinazolinone derivative and regenerating the active palladium catalyst for the next cycle. This intricate catalytic cycle highlights the precision of modern cross-coupling chemistry in constructing complex heterocyclic frameworks.
From an impurity control perspective, this mechanism offers distinct advantages over non-catalytic thermal cyclizations. The reliance on a defined catalytic cycle minimizes the formation of random polymeric byproducts often seen in harsh acid-mediated condensations. The use of a solid CO source ensures a steady, controlled release of carbon monoxide, preventing local concentration spikes that could lead to side reactions or over-carbonylation. Furthermore, the specificity of the palladium insertion into the carbon-iodine bond ensures that the reaction proceeds exclusively at the desired position on the aromatic ring, preserving other sensitive functional groups such as halogens or nitro groups that might be present on the substrate. This high level of chemoselectivity is paramount for pharmaceutical applications where impurity profiles must be strictly controlled to meet regulatory standards for active pharmaceutical ingredients.
How to Synthesize 2-Trifluoromethyl Quinazolinone Efficiently
The practical execution of this synthesis is designed for operational ease, making it accessible for both laboratory research and pilot plant operations. The procedure involves charging a reaction vessel with the palladium catalyst, ligand, base, solid CO surrogate, and the two primary substrates in an aprotic organic solvent. The mixture is then heated to facilitate the tandem reaction sequence. Detailed standardized synthesis steps follow below to guide process implementation.
- Combine palladium catalyst, ligand, base, solid CO surrogate (TFBen), and substrates in an organic solvent.
- Heat the reaction mixture to 90°C and stir for 16 to 30 hours to ensure complete conversion.
- Filter the mixture, adsorb onto silica gel, and purify via column chromatography to isolate the final derivative.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this patented methodology translates into tangible strategic benefits regarding cost, safety, and reliability. The shift from hazardous gaseous reagents to stable solid surrogates fundamentally alters the risk profile of the manufacturing process, reducing the need for specialized high-pressure equipment and extensive safety infrastructure. This simplification of the process hardware directly correlates to lower capital expenditure and reduced operational overheads. Moreover, the use of commercially available and inexpensive starting materials like o-iodoaniline derivatives ensures a stable and continuous supply chain, mitigating the risks associated with sourcing exotic or custom-synthesized precursors. The robustness of the reaction across a wide range of substrates means that a single manufacturing platform can be adapted to produce a diverse portfolio of intermediates, enhancing asset utilization and flexibility.
- Cost Reduction in Manufacturing: The economic viability of this process is driven by the elimination of expensive pre-activated substrates and the use of a catalytic amount of palladium rather than stoichiometric reagents. By replacing toxic carbon monoxide gas with a solid surrogate, the process avoids the significant costs associated with gas handling, storage, and specialized containment systems. The high yields reported in the patent examples indicate efficient atom economy, minimizing waste disposal costs and maximizing the output per batch. Additionally, the mild reaction conditions reduce energy consumption compared to high-temperature or high-pressure alternatives, contributing to a lower overall cost of production for these valuable pharmaceutical intermediates.
- Enhanced Supply Chain Reliability: The reliance on widely available commodity chemicals such as substituted anilines and trifluoroacetimidoyl chlorides ensures that raw material sourcing is not a bottleneck. Unlike methods requiring unstable intermediates that must be synthesized immediately prior to use, the reagents in this protocol are shelf-stable and can be stocked in bulk. This stability allows for better inventory management and reduces the lead time for production scheduling. The scalability of the reaction from milligram to kilogram scales without significant loss in efficiency means that supply can be rapidly ramped up to meet clinical trial demands or commercial launch requirements without the need for extensive process re-engineering.
- Scalability and Environmental Compliance: From an environmental and safety standpoint, the avoidance of carbon monoxide gas is a major compliance advantage, simplifying permitting and reducing the regulatory burden on the manufacturing facility. The use of standard organic solvents like THF, which are easily recovered and recycled, aligns with green chemistry principles and waste reduction goals. The simplified post-treatment process, involving filtration and standard chromatography, generates less hazardous waste compared to acidic workups required by traditional methods. This cleaner process profile facilitates easier scale-up to multi-ton production levels while maintaining strict adherence to environmental, health, and safety (EHS) standards required by global regulatory bodies.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the synthesis and application of these quinazolinone derivatives. The answers are derived directly from the technical specifications and experimental data provided in the patent documentation, ensuring accuracy and relevance for industry professionals evaluating this technology for their supply chains.
Q: Why is the trifluoromethyl group significant in quinazolinone drug design?
A: Introducing a trifluoromethyl group significantly enhances physicochemical properties such as metabolic stability, lipophilicity, and bioavailability, which are critical for optimizing drug candidates like anticonvulsants and antitumor agents.
Q: What safety advantages does this synthesis method offer over traditional carbonylation?
A: This method utilizes 1,3,5-tricarboxylate phenol ester (TFBen) as a solid carbon monoxide surrogate, effectively eliminating the need for handling toxic and hazardous carbon monoxide gas directly.
Q: What is the substrate scope of this palladium-catalyzed reaction?
A: The reaction demonstrates excellent compatibility with various substituents on both the aniline and imidoyl chloride components, including halogens, alkyl groups, and nitro groups, allowing for diverse structural modifications.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Quinazolinone Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in the success of drug development pipelines. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet your volume requirements with consistency and precision. We are committed to delivering products that meet stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation. Our expertise in palladium-catalyzed transformations allows us to optimize this specific synthesis route for maximum efficiency and cost-effectiveness, providing you with a competitive edge in the marketplace.
We invite you to collaborate with us to leverage this advanced synthetic technology for your projects. Please contact our technical procurement team to request a Customized Cost-Saving Analysis tailored to your specific volume needs. We are ready to provide specific COA data and comprehensive route feasibility assessments to demonstrate how our manufacturing capabilities can support your long-term supply goals. Let us be your partner in bringing innovative therapeutic solutions to market faster and more efficiently.