Advanced Manufacturing of Tezacaftor Intermediates via Optimized Reductive Cyclization
Advanced Manufacturing of Tezacaftor Intermediates via Optimized Reductive Cyclization
The pharmaceutical landscape for Cystic Fibrosis treatment has been significantly advanced by the development of CFTR modulators, with Tezacaftor standing out as a critical component in combination therapies. Patent CN111763198B discloses a groundbreaking preparation method for 5-substituted cyclopropyl formylaminoindole derivatives, specifically targeting the efficient synthesis of Tezacaftor. This technical disclosure represents a paradigm shift from complex, metal-heavy synthetic pathways to a more streamlined, cost-effective approach utilizing ammonia substitution and reductive cyclization. For R&D directors and procurement specialists, understanding this novel route is essential for securing a reliable pharmaceutical intermediates supplier capable of delivering high-purity materials at scale. The structural complexity of Tezacaftor, characterized by its difluorobenzo[d][1,3]dioxole and indole moieties, demands precise chemical engineering to ensure batch-to-batch consistency.
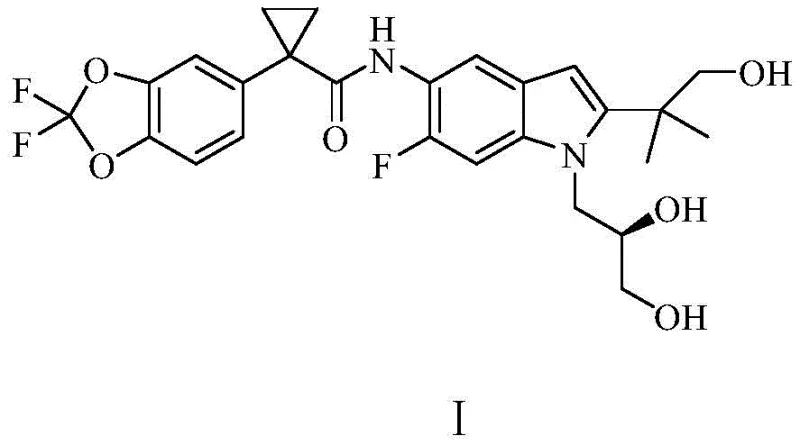
This patent outlines a robust six-step sequence that begins with readily available halogenated phenylacetonitriles and culminates in the final active pharmaceutical ingredient precursor. By focusing on inherent functional group reactivity, the inventors have designed a route that maximizes atom economy and minimizes environmental impact. The strategic use of catalytic hydrogenation not only reduces nitro groups but also facilitates the critical cyclization event that constructs the indole core, a transformation that is often the bottleneck in traditional syntheses. This report analyzes the technical merits of this invention, providing deep insights into its mechanistic advantages and commercial viability for global supply chains.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art methodologies, such as those described in US20180265501A1 and CN103038214A, rely heavily on palladium-catalyzed cross-coupling reactions to construct the molecular framework. These conventional routes typically initiate with 5-bromo-2,2-difluoro-1,3-benzodioxole, undergoing coupling with ethyl cyanoacetate in the presence of expensive catalysts like Pd(dba)2 and ligands such as tert-butylphosphine. The subsequent steps involve decarboxylation, cyclopropanation using strong bases, and multiple protection-deprotection sequences involving benzyl groups. This linear approach results in a lengthy synthesis with poor atom economy, generating significant amounts of waste salt and requiring rigorous purification to remove trace heavy metals. Furthermore, the reliance on precious metal catalysts introduces substantial cost volatility and supply chain risks, making cost reduction in pharmaceutical intermediates manufacturing difficult to achieve consistently.
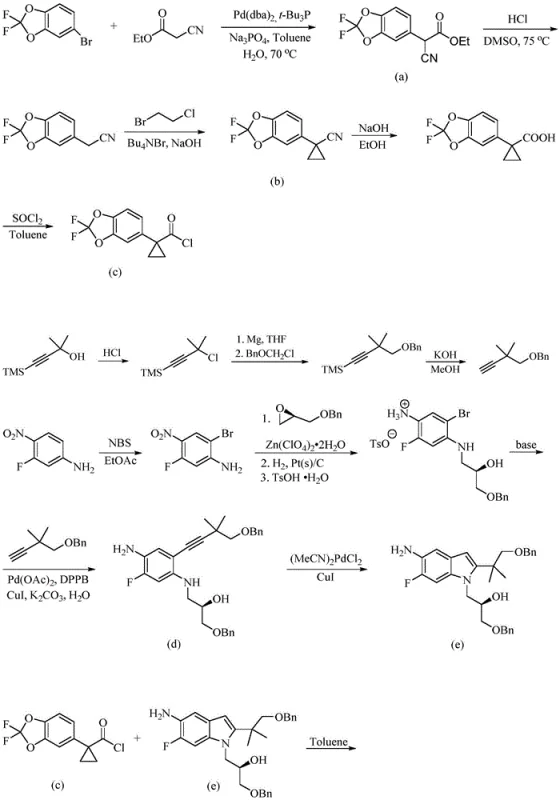
The Novel Approach
In stark contrast, the method disclosed in CN111763198B adopts a convergent strategy that drastically simplifies the synthetic tree. The novel approach utilizes 2-nitro-4-fluoro-5-halophenylacetonitrile as a key building block, leveraging the activating effect of the nitro group to facilitate nucleophilic aromatic substitution with ammonia. This eliminates the need for early-stage palladium coupling. The route proceeds through a logical sequence of amidation, dehydration condensation, and a pivotal reductive cyclization-elimination cascade. By constructing the indole ring via hydrogenation of a nitro-alkene precursor, the process avoids the harsh conditions associated with traditional indole synthesis. The final steps involve a stereoselective ring-opening with a chiral epoxide and a clean hydrogenolysis to reveal the diol functionality. This streamlined pathway not only shortens the overall reaction time but also enhances the safety profile by avoiding hazardous reagents and extreme conditions.
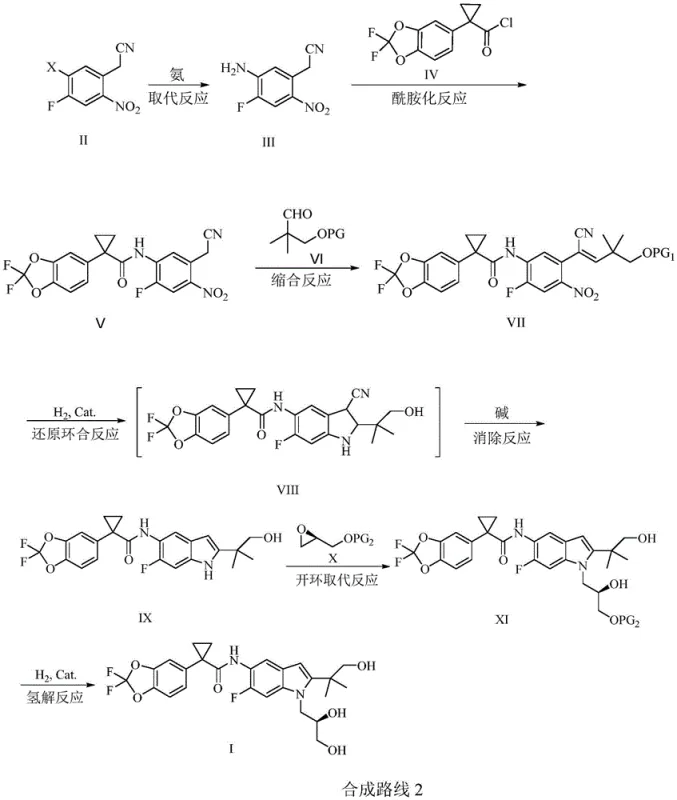
Mechanistic Insights into Reductive Cyclization and Elimination
The cornerstone of this innovative synthesis is the transformation of the nitro-alkene intermediate (Formula VII) into the indole core (Formula IX). This step involves a sophisticated tandem sequence initiated by catalytic hydrogenation. Under hydrogen pressure (0.1-0.8 MPa) and in the presence of a palladium-carbon or Raney nickel catalyst, the nitro group is selectively reduced to an amine. This newly formed amino group, positioned ortho to the electron-deficient alkene system activated by the nitrile group, undergoes an intramolecular Michael-type addition. This cyclization event forms the dihydroindole intermediate (Formula VIII). The elegance of this mechanism lies in its subsequent spontaneous or base-promoted elimination. Upon treatment with a mild base such as potassium carbonate or sodium carbonate, the intermediate eliminates hydrogen cyanide (HCN), driving the aromatization of the five-membered ring to yield the stable indole structure. This cascade reaction is highly atom-economical and demonstrates exceptional chemoselectivity, leaving other sensitive functional groups like the cyclopropane and acetal moieties intact.
Impurity control is rigorously managed through the specific choice of reaction conditions and work-up procedures. The initial ammonia substitution step is optimized to prevent over-reaction or solvent participation, ensuring high purity of the amino intermediate (Formula III). During the reductive cyclization, the use of specific solvents like methanol or ethanol helps solubilize intermediates while maintaining catalyst activity. The elimination step is carefully monitored to ensure complete aromatization without degrading the sensitive side chains. Furthermore, the patent describes effective recovery of cyanide byproducts from the filtrate, which can be recycled to prepare starting materials, thereby closing the loop on waste generation. This attention to mechanistic detail ensures that the final product meets stringent purity specifications required for regulatory approval, minimizing the burden on downstream purification processes.
How to Synthesize Tezacaftor Efficiently
The synthesis of Tezacaftor via this patented route offers a practical blueprint for laboratory and pilot-scale production. The process is divided into distinct operational units: substitution, amidation, condensation, cyclization, alkylation, and deprotection. Each step utilizes common industrial solvents and reagents, facilitating easy technology transfer. The detailed standardized synthesis steps below outline the specific parameters for temperature, pressure, and stoichiometry required to achieve optimal yields and purity.
- Perform ammonia substitution on 2-nitro-4-fluoro-5-halophenylacetonitrile to obtain the amino intermediate.
- Conduct amidation with 1-(2,2-difluoro-1,3-benzodioxazol-5-yl)cyclopropanecarbonyl chloride.
- Execute dehydration condensation with a protected aldehyde to form the alkene intermediate.
- Carry out catalytic hydrogenation for reductive cyclization followed by base-mediated elimination to form the indole core.
- Perform ring-opening substitution with a chiral epoxide and final catalytic hydrogenolysis to remove protecting groups.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this novel synthesis route presents compelling economic and logistical benefits. The primary advantage lies in the drastic simplification of the raw material portfolio. By replacing expensive brominated benzodioxoles and palladium catalysts with commodity chemicals like halogenated nitrobenzenes and ammonia, the direct material costs are significantly reduced. This shift mitigates exposure to the volatile pricing of precious metals and specialized ligands. Additionally, the reduction in reaction steps translates to shorter manufacturing lead times, allowing for faster response to market demand fluctuations. The simplified work-up procedures, which often involve simple filtration and crystallization rather than complex chromatography, further enhance throughput and reduce labor costs associated with production.
- Cost Reduction in Manufacturing: The elimination of palladium catalysts in the early stages of synthesis removes the need for expensive metal scavenging and recovery processes, which are both capital and operationally intensive. The use of cheaper starting materials like 2-nitro-4-fluoro-5-chlorophenylacetonitrile instead of brominated analogs further drives down the cost of goods sold. Moreover, the high atom economy of the reductive cyclization step means less raw material is wasted as byproduct, maximizing the yield per kilogram of input. This efficiency allows for substantial cost savings that can be passed down the supply chain or reinvested in quality assurance.
- Enhanced Supply Chain Reliability: The reliance on widely available bulk chemicals ensures a stable supply of raw materials, reducing the risk of production stoppages due to sourcing issues. The robustness of the reaction conditions, which tolerate a range of temperatures and pressures without compromising yield, adds a layer of operational resilience. This reliability is crucial for maintaining continuous production schedules and meeting the strict delivery timelines demanded by pharmaceutical clients. The ability to recycle cyanide byproducts back into the synthesis of starting materials also creates a more self-sufficient and secure manufacturing ecosystem.
- Scalability and Environmental Compliance: The process is explicitly designed for industrial scale-up, utilizing standard reactor types and avoiding hazardous reagents that require special handling permits. The generation of waste acid and wastewater is minimized compared to traditional routes, aligning with increasingly strict environmental regulations. The final purification steps involve crystallization, which is easily scalable and produces high-purity solids suitable for direct formulation. This environmental compatibility not only reduces disposal costs but also enhances the corporate sustainability profile of the manufacturing partner.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the patented synthesis of Tezacaftor. These answers are derived directly from the experimental data and beneficial effects described in the patent documentation, providing clarity on the feasibility and advantages of this method for potential partners and stakeholders.
Q: How does this new synthesis route improve upon conventional methods for Tezacaftor?
A: Unlike prior art which relies on expensive palladium couplings and long reaction sequences, this method utilizes cheap raw materials like halo-nitriles and avoids transition metal catalysts in early steps, significantly reducing cost and complexity while improving atom economy.
Q: What mechanisms ensure high purity in the reductive cyclization step?
A: The process leverages the specific activation of the nitro group for reduction and subsequent intramolecular cyclization. The selectivity is controlled by precise temperature and pressure parameters during hydrogenation, followed by a specific base-mediated elimination that aromatizes the indole ring efficiently.
Q: Is this manufacturing process suitable for large-scale industrial production?
A: Yes, the patent explicitly highlights suitability for industrial production due to simple operation, safe reaction conditions, easy work-up procedures like filtration and crystallization, and the generation of minimal waste acid and water compared to traditional routes.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Tezacaftor Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient and scalable synthesis routes for complex pharmaceutical intermediates like Tezacaftor. Our team of expert chemists has thoroughly analyzed the methodology disclosed in CN111763198B and is fully equipped to implement this advanced technology. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with precision and consistency. Our state-of-the-art facilities are designed to handle the specific requirements of reductive cyclization and catalytic hydrogenation, adhering to stringent purity specifications and rigorous QC labs to guarantee product quality that exceeds industry standards.
We invite you to collaborate with us to leverage these technological advancements for your supply chain. By partnering with us, you gain access to a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage you to contact our technical procurement team to request specific COA data and route feasibility assessments. Let us demonstrate how our commitment to innovation and quality can drive value and reliability in your Tezacaftor supply chain.