Scalable Synthesis of Chiral 2-Hydroxy-1,4-Dicarbonyl Compounds and Pantolactone Using Novel Tetrapeptide Catalysts
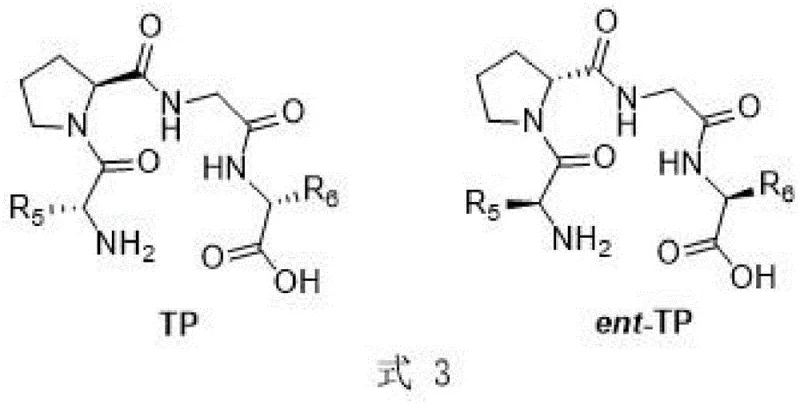
The pharmaceutical and fine chemical industries are constantly seeking robust methodologies for the construction of chiral building blocks, particularly those serving as precursors to vital vitamins and bioactive molecules. Patent CN111848320A introduces a groundbreaking advancement in this domain by disclosing a highly efficient asymmetric Aldol reaction catalyzed by novel tetrapeptides, specifically designed to synthesize chiral 2-hydroxy-1,4-dicarbonyl compounds with unprecedented stereocontrol. This technology addresses a critical bottleneck in the production of optically active pantolactone, a key intermediate for Pantothenic Acid (Vitamin B5), by overcoming the historical limitations of low enantioselectivity found in earlier organocatalytic systems. The invention demonstrates that by utilizing specific tetrapeptide sequences, designated as TP or their enantiomers ent-TP, manufacturers can achieve yields up to 99% and enantiomeric excess (ee) values reaching 99%, a significant leap from the sub-80% ee ceilings of previous methods. This breakthrough not only enhances the purity profile of the final active pharmaceutical ingredients but also streamlines the synthetic route by eliminating the need for complex resolution steps often required with inferior catalysts.
Historically, the synthesis of chiral 2-hydroxy-1,4-dicarbonyl compounds via asymmetric Aldol reactions has relied heavily on simple amino acids such as proline, histidine, or isoleucine. While these natural catalysts offered a green alternative to metal-based systems, they suffered from significant performance deficits that hindered their industrial viability. For instance, proline-catalyzed reactions of isobutyraldehyde with glyoxylates historically yielded ee values of merely 42%, while optimizations with histidine and isoleucine managed to push this boundary only to approximately 79% under specific solvent conditions. Such moderate stereoselectivity necessitates extensive and costly downstream purification processes, such as recrystallization or chromatography, to meet the stringent purity specifications required for pharmaceutical applications. Furthermore, many of these traditional protocols require high catalyst loadings or harsh reaction conditions that can compromise the stability of sensitive functional groups, thereby limiting the substrate scope and overall process efficiency for complex molecule synthesis.
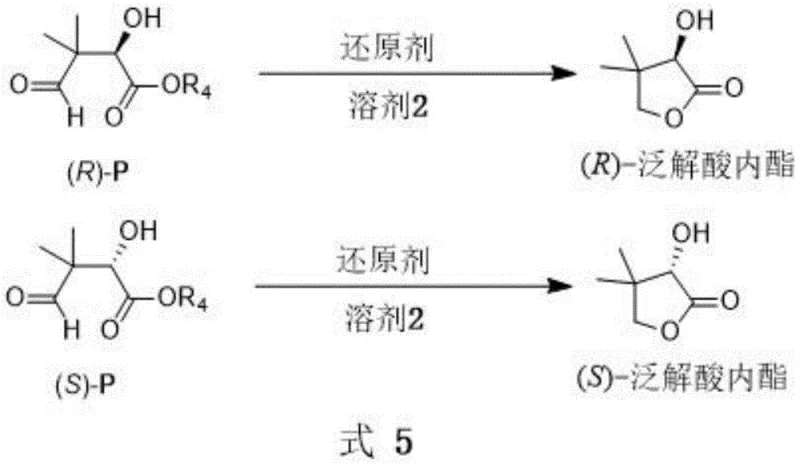
In stark contrast, the novel approach detailed in the patent leverages the structural complexity of tetrapeptides to create a highly defined chiral environment that mimics the active sites of natural enzymes. By employing catalysts such as D-Val-L-Pro-Gly-D-Leu (TP-1) or its enantiomeric counterparts, the reaction achieves superior facial selectivity during the carbon-carbon bond formation. This method allows for the flexible synthesis of both (R)- and (S)-configured products simply by switching between the TP and ent-TP catalyst series, providing a versatile platform for accessing diverse chiral scaffolds. The reaction proceeds under remarkably mild conditions, often at room temperature or with simple ice-bath cooling, utilizing common organic solvents like acetonitrile or dichloromethane. This operational simplicity, combined with the ability to tolerate a wide range of aliphatic aldehydes and glyoxylates, positions this tetrapeptide-catalyzed Aldol reaction as a superior alternative for the cost reduction in pharmaceutical intermediate manufacturing, offering a direct path to high-value chiral intermediates without the baggage of transition metal contamination.
The mechanistic elegance of this transformation lies in the unique ability of the tetrapeptide backbone to organize the transition state through a network of hydrogen bonding and steric interactions. Unlike small molecule organocatalysts that may lack sufficient structural rigidity, the tetrapeptide catalysts TP and ent-TP possess a defined secondary structure that effectively shields one face of the electrophile, directing the nucleophilic attack of the enamine or enolate intermediate with high precision. The catalyst structure, characterized by specific amino acid residues at the N-terminus and C-terminus (denoted as R5 and R6 in the patent formulas), plays a pivotal role in tuning the electronic and steric properties of the active site. This biomimetic approach ensures that the reaction maintains high fidelity even with bulky or electronically diverse substrates, such as aryl-substituted acyl formaldehydes or cyclic aliphatic aldehydes. The result is a catalytic cycle that minimizes the formation of unwanted diastereomers and enantiomers, thereby simplifying the impurity profile and enhancing the overall atom economy of the process.
Furthermore, the robustness of these peptide-based catalysts contributes significantly to the reproducibility of the synthesis. The patent data indicates that the catalysts remain stable under the reaction conditions, avoiding the rapid decomposition or racemization often observed with more fragile enzymatic systems. This stability allows for consistent performance across different batches, a critical factor for supply chain reliability. The broad substrate scope demonstrated in the examples, covering linear alkyl chains, cycloalkyl groups, and various aromatic substitutions, underscores the versatility of this methodology. Whether synthesizing precursors for pantolactone or other complex polyketide-like structures, the tetrapeptide catalyst provides a reliable foundation for constructing quaternary carbon centers with adjacent hydroxyl groups, a motif frequently encountered in bioactive natural products and drug candidates.
How to Synthesize Chiral 2-Hydroxy-1,4-Dicarbonyl Compounds Efficiently
The practical implementation of this technology involves a straightforward protocol that integrates seamlessly into existing laboratory and pilot plant workflows. The process begins with the dissolution of the precise amount of tetrapeptide catalyst in a selected solvent, followed by the sequential addition of the aldehyde and the glyoxylate or acyl formaldehyde component. Reaction monitoring is typically conducted via thin-layer chromatography (TLC) using standard visualization techniques, ensuring that the reaction is quenched at the optimal conversion point to maximize yield and purity. Following the reaction, a standard aqueous workup involving extraction and drying steps isolates the crude product, which can then be purified via column chromatography if necessary, although the high selectivity often minimizes the burden on purification resources. This streamlined procedure highlights the ease of operation and the potential for rapid technology transfer from bench scale to commercial production.
- Dissolve the tetrapeptide catalyst (TP or ent-TP, 0.025 mmol) in an appropriate solvent such as acetonitrile or dichloromethane within a reaction vessel.
- Add the aliphatic aldehyde substrate (1.0 mmol) to the mixture under stirring, typically maintaining the temperature in an ice-water bath initially before allowing it to warm to room temperature.
- Introduce the electrophile, either glyoxylate or acyl formaldehyde monohydrate (0.5 mmol), and stir until completion as monitored by TLC, followed by standard aqueous workup and purification.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this tetrapeptide-catalyzed synthesis offers compelling strategic advantages that extend beyond mere technical performance. The elimination of transition metals from the catalytic cycle is a primary driver for cost reduction in manufacturing, as it removes the necessity for expensive and time-consuming heavy metal scavenging steps that are mandatory for compliance with strict regulatory limits on residual metals in pharmaceutical products. By utilizing an organocatalytic system based on amino acid derivatives, the process inherently aligns with green chemistry principles, reducing the environmental footprint associated with waste disposal and solvent usage. This alignment not only mitigates regulatory risks but also enhances the sustainability profile of the supply chain, a factor increasingly weighted in vendor selection criteria by major multinational corporations.
- Cost Reduction in Manufacturing: The high enantioselectivity achieved by this method drastically reduces the material loss associated with chiral resolution or recycling of unwanted isomers. In traditional processes where ee values hover around 60-70%, nearly half of the produced material may be discarded or require energy-intensive reprocessing. By consistently delivering ee values above 95% and often reaching 99%, this technology maximizes the yield of the desired isomer, directly translating to substantial cost savings in raw material consumption and processing time. Additionally, the mild reaction conditions reduce energy expenditures related to heating or cryogenic cooling, further optimizing the operational expenditure (OPEX) profile of the manufacturing process.
- Enhanced Supply Chain Reliability: The reliance on stable, synthetically accessible tetrapeptide catalysts ensures a secure supply of critical reagents, unlike biocatalytic methods that may suffer from batch-to-batch variability or shelf-life issues. The robustness of the chemical catalysts means that inventory management is simplified, and production schedules are less prone to disruption due to catalyst degradation. Furthermore, the use of common, commodity-grade solvents such as acetonitrile and ethyl acetate ensures that the process is not vulnerable to supply shocks associated with exotic or highly specialized reagents, thereby guaranteeing continuity of supply for high-purity pharmaceutical intermediates.
- Scalability and Environmental Compliance: The simplicity of the workup procedure and the absence of toxic metal residues facilitate easier scale-up from kilogram to multi-ton quantities. This scalability is crucial for meeting the demands of global markets without the need for specialized containment equipment required for hazardous chemistries. From an environmental compliance perspective, the reduced generation of hazardous waste and the use of biodegradable peptide-based catalysts support corporate sustainability goals. This makes the technology particularly attractive for companies aiming to reduce their carbon footprint and adhere to increasingly stringent environmental regulations governing chemical manufacturing.
Frequently Asked Questions (FAQ)
To assist technical teams in evaluating the feasibility of integrating this technology into their existing portfolios, we have compiled answers to common inquiries regarding the operational parameters and scope of the tetrapeptide-catalyzed Aldol reaction. These insights are derived directly from the experimental data and technical specifications outlined in the patent documentation, providing a clear picture of what can be expected during process development and scale-up activities.
Q: What represents the primary advantage of tetrapeptide catalysts over traditional amino acid catalysts in this synthesis?
A: Traditional catalysts like proline or histidine typically achieve enantiomeric excess (ee) values below 80%, whereas the tetrapeptide catalysts disclosed in CN111848320A consistently deliver ee values up to 99%, significantly reducing the need for downstream purification.
Q: Can this method produce both enantiomers of the target chiral compounds?
A: Yes, the patent describes the use of both the tetrapeptide TP and its enantiomer ent-TP, allowing for the selective synthesis of either the (R)-configured or (S)-configured 2-hydroxy-1,4-dicarbonyl compounds depending on the specific catalyst employed.
Q: Is this process suitable for large-scale manufacturing of pantolactone?
A: The process utilizes mild reaction conditions, commercially available solvents, and avoids toxic heavy metals, making it highly amenable to commercial scale-up for the production of high-purity pantolactone intermediates.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Chiral 2-Hydroxy-1,4-Dicarbonyl Compounds Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of advanced organocatalytic technologies like the one described in CN111848320A for the production of high-value chiral intermediates. As a leading CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical benefits of this tetrapeptide-catalyzed route are fully realized in a GMP-compliant manufacturing environment. Our rigorous QC labs and stringent purity specifications guarantee that every batch of chiral 2-hydroxy-1,4-dicarbonyl compounds or pantolactone derivatives meets the exacting standards required by the global pharmaceutical industry, providing our partners with the confidence needed to accelerate their drug development timelines.
We invite you to engage with our technical procurement team to discuss how this innovative synthesis route can be tailored to your specific project needs. By requesting a Customized Cost-Saving Analysis, you can gain a deeper understanding of the economic benefits associated with switching to this metal-free, high-selectivity process. We are prepared to provide specific COA data and route feasibility assessments to demonstrate how our capabilities align with your goals for cost reduction in pharmaceutical intermediate manufacturing and supply chain optimization.