Advanced Pd-Catalyzed Carbonylation for Scalable Production of 2-Trifluoromethyl Quinazolinone Derivatives
Advanced Pd-Catalyzed Carbonylation for Scalable Production of 2-Trifluoromethyl Quinazolinone Derivatives
The pharmaceutical industry continuously seeks robust synthetic methodologies for constructing privileged scaffolds that exhibit potent biological activities. Patent CN112125856A discloses a groundbreaking preparation method for 2-trifluoromethyl substituted quinazolinone derivatives, a class of compounds renowned for their diverse pharmacological profiles ranging from anticancer to anticonvulsant properties. As illustrated in the structural diversity of known bioactive molecules, the quinazolinone core is a critical motif in modern drug discovery. 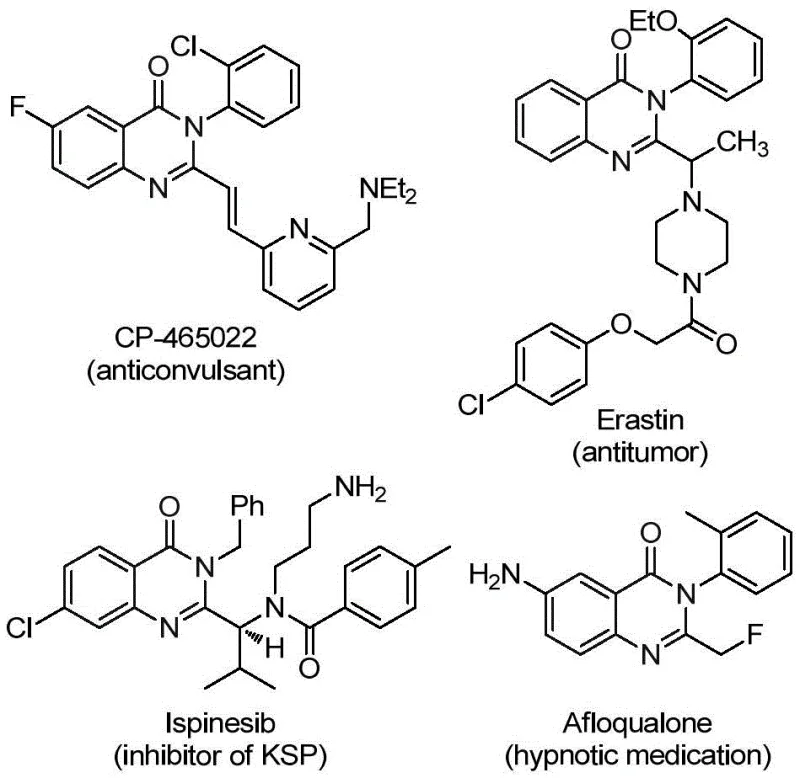 This novel approach leverages a transition metal palladium-catalyzed carbonylation tandem reaction, utilizing readily available o-iodoaniline and trifluoroethylimidoyl chloride as starting materials. By employing a solid carbon monoxide substitute, the process not only enhances operational safety but also streamlines the synthetic workflow, making it an attractive candidate for industrial scale-up and reliable pharmaceutical intermediate supplier operations.
This novel approach leverages a transition metal palladium-catalyzed carbonylation tandem reaction, utilizing readily available o-iodoaniline and trifluoroethylimidoyl chloride as starting materials. By employing a solid carbon monoxide substitute, the process not only enhances operational safety but also streamlines the synthetic workflow, making it an attractive candidate for industrial scale-up and reliable pharmaceutical intermediate supplier operations.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of 2-trifluoromethyl substituted quinazolinone derivatives has been fraught with significant technical and safety challenges that hinder efficient manufacturing. Traditional routes often rely on the cyclization of anthranilamides with ethyl trifluoroacetate or trifluoroacetic anhydride, which frequently necessitate harsh reaction conditions and expensive, pre-activated substrates. Furthermore, many established protocols involve the direct use of carbon monoxide gas, a highly toxic and colorless substance that poses severe safety risks in large-scale production environments. These conventional methods also suffer from narrow substrate scopes, limiting the ability to introduce diverse functional groups required for structure-activity relationship (SAR) studies. Consequently, the low yields and difficult purification processes associated with these older techniques result in increased production costs and extended lead times, creating bottlenecks for cost reduction in API manufacturing.
The Novel Approach
In stark contrast, the methodology described in patent CN112125856A offers a transformative solution by utilizing a palladium-catalyzed carbonylation strategy that circumvents the drawbacks of prior art. This innovative route employs 1,3,5-tricarboxylate phenol ester (TFBen) as a solid surrogate for carbon monoxide, thereby eliminating the hazards associated with gaseous CO while maintaining high reaction efficiency. The process demonstrates exceptional compatibility with a wide array of substituents, allowing for the facile synthesis of derivatives with varying electronic and steric properties. 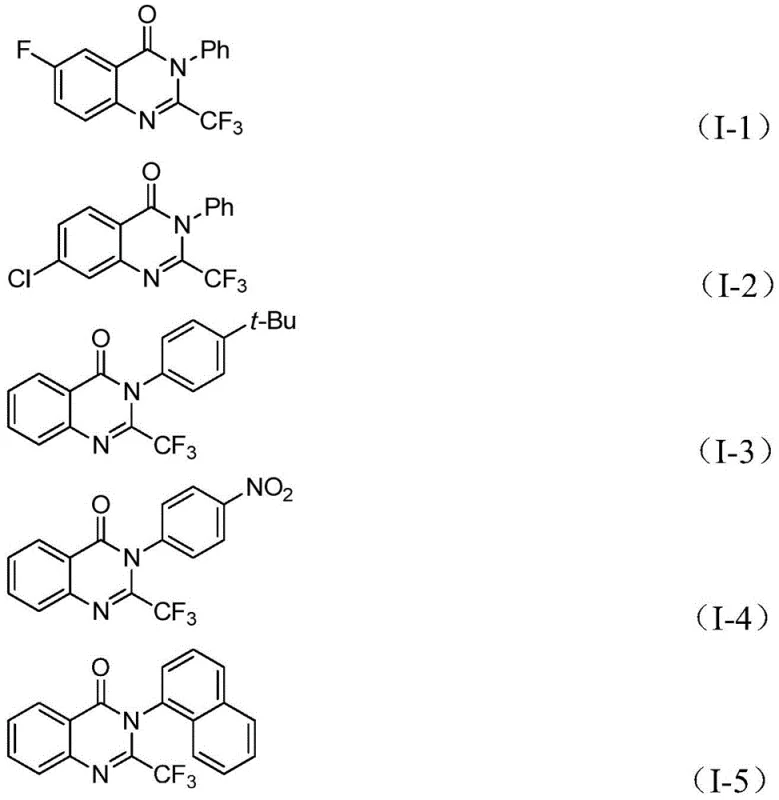 As shown in the specific examples, the method successfully produces complex structures such as those with nitro, tert-butyl, and naphthyl groups with commendable yields. This versatility ensures that research and development teams can rapidly access high-purity quinazolinone derivatives for screening, while procurement teams benefit from a simplified supply chain driven by cheap and easily obtainable starting materials.
As shown in the specific examples, the method successfully produces complex structures such as those with nitro, tert-butyl, and naphthyl groups with commendable yields. This versatility ensures that research and development teams can rapidly access high-purity quinazolinone derivatives for screening, while procurement teams benefit from a simplified supply chain driven by cheap and easily obtainable starting materials.
Mechanistic Insights into Palladium-Catalyzed Carbonylation Cyclization
The success of this synthetic route lies in its elegant catalytic cycle, which orchestrates multiple bond-forming events in a single pot. The reaction likely initiates with a base-promoted intermolecular carbon-nitrogen bond coupling between the o-iodoaniline and the trifluoroethylimidoyl chloride, generating a trifluoroacetamidine intermediate. Subsequently, the palladium catalyst inserts into the carbon-iodine bond to form a divalent palladium species. Under heating conditions, the solid CO source TFBen decomposes to release carbon monoxide in situ, which then inserts into the carbon-palladium bond to create an acyl palladium intermediate. This sequence is critical for introducing the carbonyl functionality essential for the quinazolinone ring closure.
Following the CO insertion, the base facilitates the formation of a palladium-nitrogen bond, leading to the generation of a seven-membered ring palladium intermediate. The final step involves a reductive elimination that releases the desired 2-trifluoromethyl substituted quinazolinone derivative and regenerates the active palladium catalyst. 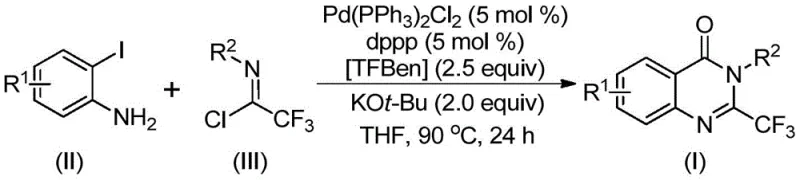 This mechanistic pathway not only explains the high efficiency observed but also highlights the impurity control mechanisms inherent in the design. By avoiding unstable intermediates and harsh acidic conditions often found in traditional cyclizations, the process minimizes side reactions and degradation, ensuring a cleaner crude product profile that simplifies downstream purification and enhances the overall quality of the high-purity pharmaceutical intermediates produced.
This mechanistic pathway not only explains the high efficiency observed but also highlights the impurity control mechanisms inherent in the design. By avoiding unstable intermediates and harsh acidic conditions often found in traditional cyclizations, the process minimizes side reactions and degradation, ensuring a cleaner crude product profile that simplifies downstream purification and enhances the overall quality of the high-purity pharmaceutical intermediates produced.
How to Synthesize 2-Trifluoromethyl Quinazolinone Efficiently
The operational simplicity of this protocol makes it highly suitable for both laboratory optimization and commercial production. The procedure involves charging a reactor with the palladium catalyst, ligand, base, solid CO source, and substrates in an aprotic solvent such as tetrahydrofuran. The mixture is then heated to 90°C for a defined period, typically between 16 to 30 hours, to ensure complete conversion. Detailed standardized synthesis steps are provided in the guide below to assist technical teams in replicating these results with precision.
- Combine palladium catalyst, dppp ligand, potassium tert-butoxide, TFBen, trifluoroethylimidoyl chloride, and o-iodoaniline in an organic solvent like THF.
- Heat the reaction mixture to 90°C and maintain stirring for 16 to 30 hours to allow the carbonylation cyclization to proceed.
- Upon completion, filter the mixture, mix with silica gel, and purify via column chromatography to isolate the target derivative.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this patented methodology offers substantial strategic benefits that extend beyond mere chemical yield. The shift from hazardous gaseous reagents to stable solid surrogates fundamentally alters the risk profile of the manufacturing process, reducing the need for specialized gas handling infrastructure and lowering insurance and compliance costs. Furthermore, the use of commercially available and inexpensive starting materials like o-iodoaniline derivatives ensures a stable and resilient supply chain, mitigating the risks associated with sourcing exotic or custom-synthesized precursors. This stability is crucial for maintaining continuous production schedules and meeting the demanding delivery timelines of global pharmaceutical clients.
- Cost Reduction in Manufacturing: The economic viability of this process is significantly enhanced by the elimination of expensive pre-activated substrates and the avoidance of complex gas handling systems. By utilizing cheap and readily available reagents, the overall material cost is drastically reduced, allowing for more competitive pricing strategies. Additionally, the high conversion rates and clean reaction profiles minimize waste generation and solvent consumption, contributing to further operational savings. These factors collectively drive down the cost of goods sold (COGS), enabling significant cost reduction in API manufacturing without compromising on quality or purity standards.
- Enhanced Supply Chain Reliability: The reliance on commodity chemicals rather than specialized reagents ensures that raw material availability is not a bottleneck for production. Since the key components such as the palladium catalyst and the solid CO source are stable and have long shelf lives, inventory management becomes more predictable and less prone to disruption. This reliability translates into shorter lead times for high-purity pharmaceutical intermediates, allowing customers to accelerate their own drug development pipelines. The robust nature of the reaction conditions also means that the process is less sensitive to minor variations in raw material quality, further stabilizing the supply chain.
- Scalability and Environmental Compliance: From an environmental and regulatory perspective, this method aligns perfectly with green chemistry principles by avoiding toxic gases and reducing hazardous waste. The use of a solid CO source simplifies the engineering controls required for scale-up, making the transition from kilogram to ton-scale production smoother and safer. This ease of commercial scale-up of complex heterocycles ensures that manufacturers can rapidly respond to increased market demand. Moreover, the reduced environmental footprint facilitates easier regulatory approval and compliance with increasingly stringent global environmental standards, safeguarding the long-term sustainability of the manufacturing operation.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding this synthesis technology, derived directly from the patent specifications and experimental data. Understanding these details is essential for evaluating the feasibility of integrating this route into existing production workflows. The answers provided reflect the specific advantages and operational parameters defined in the intellectual property, ensuring accuracy and relevance for technical decision-makers.
Q: What is the primary safety advantage of this synthesis method compared to traditional carbonylation?
A: This method utilizes 1,3,5-tricarboxylate phenol ester (TFBen) as a solid carbon monoxide substitute, effectively eliminating the need for handling toxic and hazardous carbon monoxide gas directly.
Q: Does this catalytic system support a broad range of substrate substituents?
A: Yes, the protocol demonstrates excellent compatibility with various substituents on both the o-iodoaniline and the imidoyl chloride, including halogens, alkyl groups, and nitro groups, ensuring high versatility.
Q: What are the typical reaction conditions required for this transformation?
A: The reaction typically proceeds in an aprotic solvent like THF at 90°C for 16 to 30 hours, using a palladium catalyst system with KOt-Bu as the base.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Quinazolinone Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that advanced synthetic methodologies play in accelerating drug discovery and development. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that promising laboratory results are seamlessly translated into industrial reality. We are committed to delivering high-quality intermediates that meet stringent purity specifications, supported by our rigorous QC labs and state-of-the-art analytical capabilities. By leveraging technologies like the one described in CN112125856A, we empower our partners to overcome synthetic hurdles and bring life-saving medicines to market faster.
We invite you to collaborate with us to explore the full potential of this efficient carbonylation technology for your specific project needs. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your volume requirements and quality targets. Please contact us today to request specific COA data and route feasibility assessments, and let us demonstrate how our expertise can optimize your supply chain and reduce your overall development costs.