Advanced Mo/Cu Co-Catalyzed Synthesis of 3-Trifluoromethyl-1,2,4-Triazoles for Pharmaceutical Applications
Advanced Mo/Cu Co-Catalyzed Synthesis of 3-Trifluoromethyl-1,2,4-Triazoles for Pharmaceutical Applications
The pharmaceutical industry continuously seeks robust and efficient synthetic routes for heterocyclic scaffolds that serve as the backbone of modern therapeutics. A significant breakthrough in this domain is detailed in patent CN113307778A, which discloses a novel preparation method for 3-trifluoromethyl substituted 1,2,4-triazole compounds. This class of molecules is not merely academic; it is foundational to high-value drugs such as Sitagliptin, Maraviroc, and Deferasirox, as illustrated by the molecular frameworks present in current medicinal chemistry. The introduction of a trifluoromethyl group is particularly strategic, as it drastically enhances the lipophilicity, metabolic stability, and bioavailability of the parent molecule, making these intermediates critical for next-generation drug discovery. The disclosed method leverages a unique dual-catalyst system involving molybdenum hexacarbonyl and cuprous acetate to achieve high-yield cycloaddition under remarkably mild conditions.
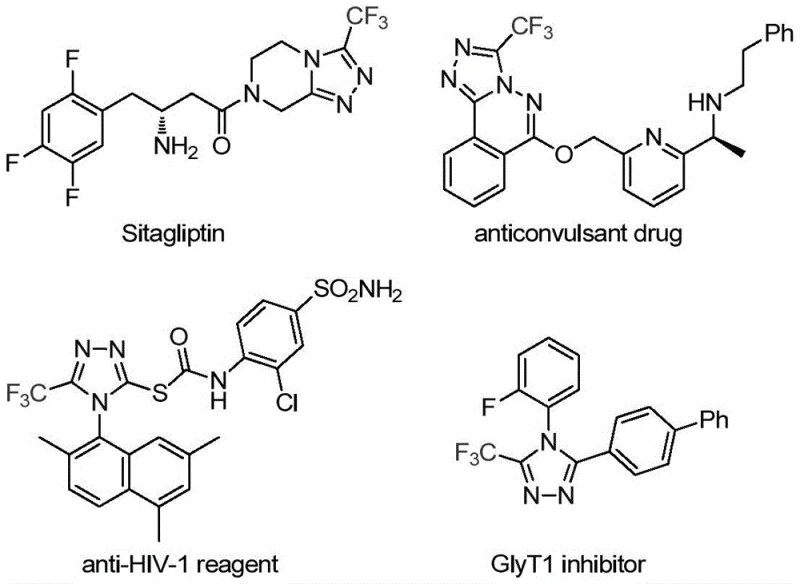
This technological advancement addresses a long-standing bottleneck in the synthesis of polysubstituted 1,2,4-triazoles. While traditional literature reports often rely on the cyclization of trifluoroacetyl hydrazine with amidines or the hydrazinolysis of oxazolinones, these pathways frequently suffer from harsh reaction conditions, limited substrate scope, or the use of hazardous reagents. The new methodology described in the patent offers a streamlined alternative by utilizing trifluoroethylimidoyl chloride and functionalized isonitriles as key building blocks. By operating at moderate temperatures between 70°C and 90°C, this process minimizes energy consumption and thermal degradation risks, positioning it as a superior candidate for the reliable supply of high-purity pharmaceutical intermediates required by global R&D teams.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of the 1,2,4-triazole ring has been fraught with synthetic challenges that impede efficient manufacturing. Conventional strategies often necessitate the use of hydrazine derivatives, which are notoriously unstable and pose significant safety hazards during storage and handling on an industrial scale. Furthermore, methods involving diazonium salts or trifluorodiazoethane require stringent safety protocols due to their explosive nature, complicating the commercial scale-up of complex polymer additives or API precursors. Many existing protocols also struggle with regioselectivity, often yielding mixtures of fully substituted triazoles rather than the specific 3,4-disubstituted isomers required for certain biological activities. Additionally, the reliance on expensive transition metal catalysts or multi-step sequences increases the overall cost of goods sold (COGS) and extends the lead time for high-purity intermediates, creating friction in the supply chain for time-sensitive drug development projects.
The Novel Approach
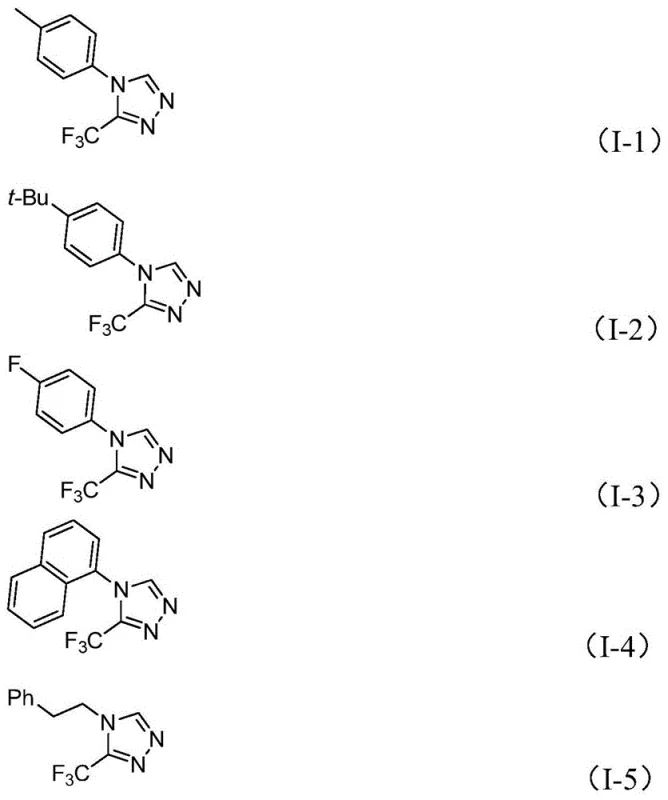
In stark contrast, the invention presented in CN113307778A introduces a paradigm shift by employing a molybdenum and copper co-catalyzed cycloaddition reaction. This approach utilizes readily available starting materials—specifically trifluoroethylimidoyl chloride and functionalized isonitriles (NIITP)—which are commercially accessible and cost-effective. The reaction design allows for the direct formation of the five-membered heterocyclic ring in a single pot, eliminating the need for isolating unstable intermediates. As demonstrated by the diverse array of products synthesized, including those with phenethyl, aryl, and naphthyl substituents, this method offers exceptional substrate tolerance. The ability to introduce various functional groups such as methyl, methoxy, fluoro, and chloro without compromising yield underscores the versatility of this route for generating focused libraries for structure-activity relationship (SAR) studies.
Mechanistic Insights into Mo/Cu Co-Catalyzed Cycloaddition
The success of this synthetic transformation lies in the synergistic interaction between the molybdenum and copper catalysts, which orchestrate a precise sequence of bond-forming events. The mechanism initiates with the activation of the functionalized isonitrile by molybdenum hexacarbonyl, forming a reactive metal-isocyanide complex. This activation lowers the energy barrier for the subsequent nucleophilic attack. Concurrently, the cuprous acetate acts as a Lewis acid promoter, facilitating the [3+2] cycloaddition between the activated isonitrile species and the trifluoroethylimidoyl chloride. This step is critical for constructing the 1,2,4-triazole core with high regioselectivity, ensuring that the trifluoromethyl group is positioned correctly at the 3-position of the ring. The final step involves the elimination of triphenylphosphine oxide, driven by the presence of water in the system or during workup, to release the final aromatic triazole product.
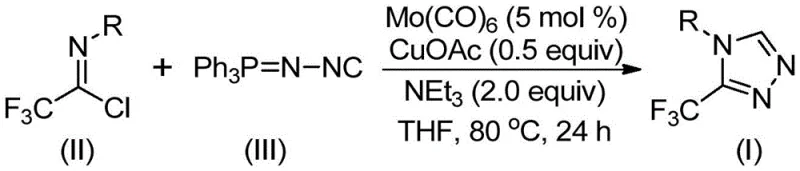
From an impurity control perspective, this mechanism offers distinct advantages for process chemistry. The use of triethylamine as a base not only neutralizes the HCl byproduct generated during the reaction but also helps maintain the catalytic cycle's efficiency. The reaction conditions are sufficiently mild to prevent the decomposition of sensitive functional groups on the aromatic rings, thereby minimizing the formation of side products such as dehalogenated species or hydrolyzed amides. Furthermore, the stoichiometry is optimized with a slight excess of the functionalized isonitrile (1.5 equivalents relative to the imidoyl chloride), which drives the reaction to completion while remaining easy to separate during the purification phase. This precise control over reaction parameters ensures a clean impurity profile, which is a paramount concern for R&D directors overseeing the development of clinical candidates.
How to Synthesize 3-Trifluoromethyl-1,2,4-Triazole Efficiently
The operational simplicity of this protocol makes it highly attractive for both laboratory discovery and pilot plant operations. The procedure involves charging a reaction vessel with the catalysts, base, and substrates in a polar aprotic solvent, followed by heating. The robustness of the system allows for a wide window of reaction times (18 to 30 hours) and temperatures (70 to 90°C), providing flexibility for process optimization. For detailed standard operating procedures regarding reagent grades, specific addition rates, and safety handling of the isonitrile species, please refer to the technical guidelines below.
- Combine molybdenum hexacarbonyl (5 mol %), cuprous acetate (0.5 equiv), triethylamine (2.0 equiv), trifluoroethylimidoyl chloride, and functionalized isonitrile in THF solvent within a reaction vessel.
- Heat the reaction mixture to a temperature range of 70-90°C and maintain stirring for a duration of 18 to 30 hours to ensure complete conversion.
- Upon completion, filter the mixture to remove solids, mix the filtrate with silica gel, and perform column chromatography purification to isolate the pure triazole product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the transition to this novel synthetic route represents a strategic opportunity to optimize the sourcing of critical heterocyclic building blocks. The shift away from hazardous hydrazine chemistry towards stable imidoyl chlorides and isonitriles significantly reduces the regulatory burden and safety infrastructure costs associated with manufacturing. This inherently safer chemistry translates to lower insurance premiums and reduced downtime for safety audits, contributing to substantial cost savings in pharmaceutical intermediate manufacturing. Moreover, the reliance on commodity chemicals like THF and triethylamine, rather than exotic or proprietary reagents, ensures a resilient supply chain that is less susceptible to market volatility or geopolitical disruptions affecting raw material availability.
- Cost Reduction in Manufacturing: The economic viability of this process is bolstered by the use of earth-abundant metals. Replacing precious metal catalysts like palladium or platinum with inexpensive cuprous acetate and molybdenum hexacarbonyl drastically lowers the catalyst cost per kilogram of product. Additionally, the high reaction efficiency and yields reported in the patent examples mean that less raw material is wasted, improving the overall mass balance. The simplified post-treatment, which involves basic filtration and standard chromatography, eliminates the need for complex extraction sequences or specialized scavenging resins, further driving down the operational expenditure (OPEX) associated with production.
- Enhanced Supply Chain Reliability: The starting materials for this synthesis, including trifluoroethylimidoyl chloride and various substituted anilines, are widely available from global chemical suppliers. This abundance ensures that production schedules can be maintained without the risk of bottlenecks caused by single-source dependencies. The scalability of the reaction, demonstrated effectively at the gram level in the patent data, suggests a smooth path to kilogram and multi-ton production. This scalability is crucial for meeting the surging demand for trifluoromethylated APIs in the treatment of diabetes and viral infections, ensuring consistent delivery to downstream formulation partners.
- Scalability and Environmental Compliance: From an environmental, health, and safety (EHS) standpoint, this method aligns well with green chemistry principles. The reaction operates in THF, a solvent that is easily recovered and recycled, minimizing liquid waste generation. The absence of heavy metal contaminants in the final product simplifies the purification process and ensures compliance with strict ICH Q3D guidelines for elemental impurities in drug substances. The mild thermal conditions also reduce the facility's carbon footprint by lowering energy consumption for heating and cooling, making this a sustainable choice for environmentally conscious manufacturing partners seeking to reduce their ecological impact.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and beneficial effects outlined in the patent documentation, providing clarity for technical teams evaluating this route for adoption.
Q: What are the primary advantages of this Mo/Cu co-catalyzed method over traditional hydrazine cyclization?
A: Unlike traditional methods that often rely on unstable hydrazines or harsh cyclization conditions, this novel approach utilizes stable trifluoroethylimidoyl chloride and functionalized isonitriles. The reaction proceeds under mild thermal conditions (70-90°C) with high atom economy and avoids the safety hazards associated with handling explosive hydrazine derivatives.
Q: Is this synthesis method suitable for large-scale commercial production?
A: Yes, the patent data explicitly demonstrates successful expansion to gram-level reactions with high efficiency. The use of inexpensive catalysts like cuprous acetate and common solvents like THF, combined with a straightforward workup procedure involving filtration and chromatography, indicates strong potential for cost-effective scale-up to kilogram or tonnage levels.
Q: What is the substrate scope for the R-group in this triazole synthesis?
A: The method exhibits excellent functional group tolerance. The R-group can be a phenethyl group or a substituted/unsubstituted aryl group. Compatible substituents include alkyl groups (methyl, ethyl, t-butyl), alkoxy groups, halogens (fluoro, chloro), and electron-withdrawing groups like nitro, allowing for the synthesis of diverse libraries for SAR studies.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3-Trifluoromethyl-1,2,4-Triazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the pivotal role that advanced heterocyclic intermediates play in accelerating drug discovery pipelines. Our team of expert process chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project transitions seamlessly from benchtop to marketplace. We are committed to delivering high-purity 3-trifluoromethyl-1,2,4-triazole derivatives that meet stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation. Whether you require custom synthesis for SAR exploration or bulk manufacturing for clinical trials, our infrastructure is designed to support your timeline and quality requirements with unwavering reliability.
We invite you to leverage our technical expertise to optimize your supply chain for these critical building blocks. By partnering with us, you gain access to a Customized Cost-Saving Analysis tailored to your specific volume needs and purity targets. We encourage you to contact our technical procurement team today to request specific COA data for our catalog compounds or to discuss route feasibility assessments for your proprietary molecules. Let us collaborate to bring your next-generation therapeutics to market faster and more efficiently.