Scalable Synthesis of Trifluoromethyl-1,2,4-Triazoles: A Metal-Free Route for High-Purity Pharmaceutical Intermediates
Scalable Synthesis of Trifluoromethyl-1,2,4-Triazoles: A Metal-Free Route for High-Purity Pharmaceutical Intermediates
The development of efficient synthetic routes for nitrogen-containing heterocycles remains a cornerstone of modern medicinal chemistry, particularly for scaffolds that exhibit potent biological activity. Patent CN113105402B discloses a groundbreaking preparation method for 3,4,5-trisubstituted 1,2,4-triazole compounds, a structural motif prevalent in high-value pharmaceuticals such as sitagliptin, maraviroc, and deferasirox. This technology addresses critical bottlenecks in the manufacturing of trifluoromethyl-substituted heterocycles by employing a metal-free, iodine-promoted cyclization strategy. The significance of this innovation lies in its ability to introduce both trifluoromethyl and acyl groups simultaneously into the triazole core, a transformation that has historically been challenging due to the electron-withdrawing nature of the CF3 group and the sensitivity of the triazole ring to harsh conditions. By leveraging a tandem sequence involving Kornblum oxidation and subsequent cyclization, this process offers a robust pathway for producing complex intermediates essential for the next generation of therapeutic agents.
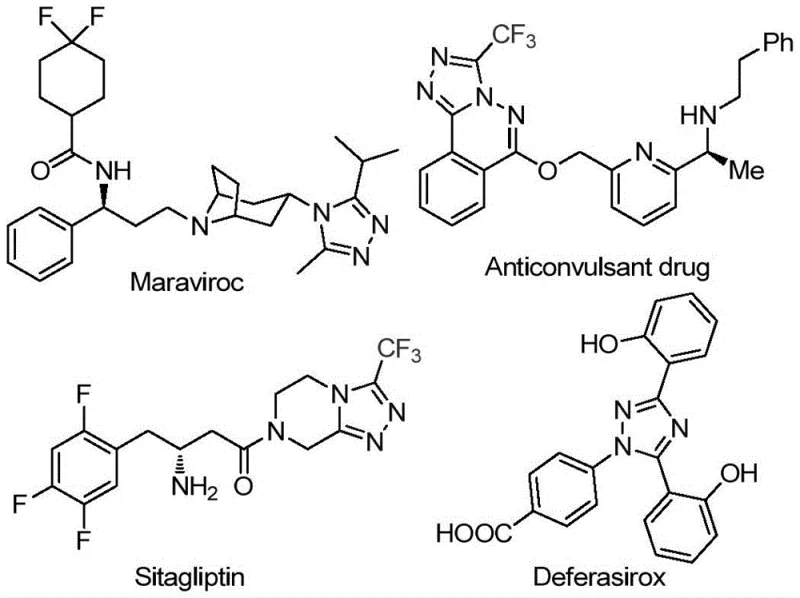
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional methodologies for constructing 1,2,4-triazole rings often rely heavily on transition metal catalysis or require extremely rigorous reaction conditions that are difficult to maintain on an industrial scale. Many existing protocols necessitate the use of expensive palladium or copper catalysts, which not only inflate the raw material costs but also introduce significant downstream purification challenges due to the stringent regulatory limits on residual heavy metals in active pharmaceutical ingredients (APIs). Furthermore, conventional routes frequently demand strictly anhydrous and oxygen-free environments, requiring specialized equipment and inert gas purging systems that increase capital expenditure and operational complexity. The introduction of trifluoromethyl groups into these heterocyclic systems is particularly problematic, as standard electrophilic fluorination reagents can be hazardous and difficult to handle, while nucleophilic approaches often suffer from poor regioselectivity and low yields. These cumulative factors result in prolonged production cycles, higher waste generation, and ultimately, a less competitive cost structure for the final pharmaceutical intermediate.
The Novel Approach
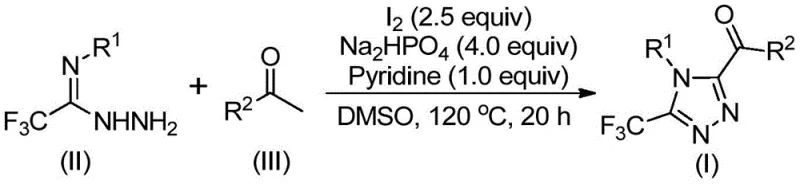
In stark contrast to these legacy methods, the novel approach detailed in the patent utilizes a simple yet highly effective iodine-promoted system in dimethyl sulfoxide (DMSO). This method bypasses the need for transition metals entirely, relying instead on the oxidative power of elemental iodine to facilitate the transformation of readily available aryl ethyl ketones into the desired triazole scaffold. The process is remarkably tolerant of ambient conditions, eliminating the necessity for glovebox techniques or extensive drying of solvents, which drastically simplifies the operational workflow. By starting from cheap and commercially abundant aryl ketones and trifluoroethylimide hydrazides, the synthesis achieves high atom economy and reduces the reliance on exotic starting materials. The versatility of this approach is demonstrated by its successful application across a wide range of substrates, including those with electron-donating and electron-withdrawing groups, proving its utility as a general platform for diversifying the triazole library for drug discovery and process development.
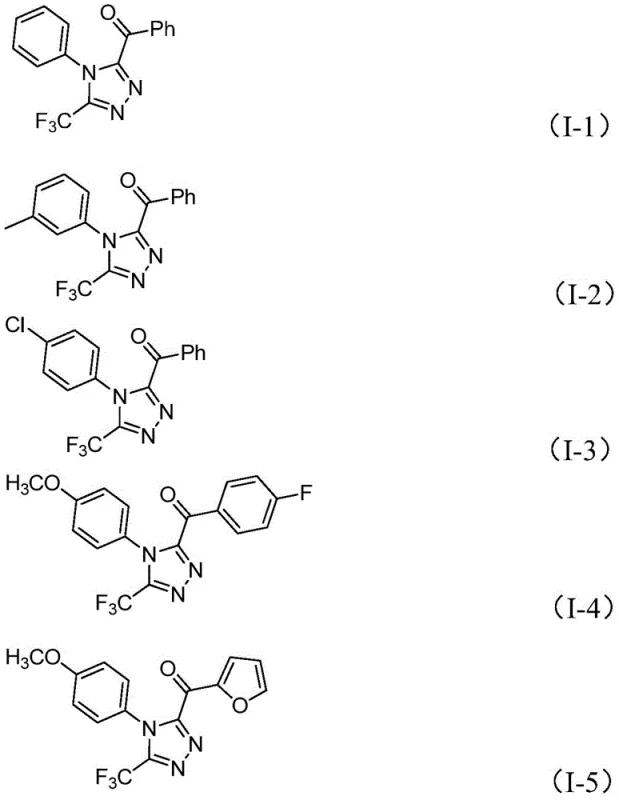
Mechanistic Insights into Iodine-Promoted Tandem Cyclization
The mechanistic pathway of this transformation is a sophisticated interplay of oxidation and condensation reactions driven by the unique properties of the iodine-DMSO system. Initially, the aryl ethyl ketone undergoes an iodination followed by a Kornblum oxidation, mediated by DMSO, to generate an aryl diketone intermediate in situ. This oxidative step is crucial as it activates the methyl group adjacent to the carbonyl, rendering it susceptible to nucleophilic attack. Subsequently, the trifluoroethylimide hydrazide acts as a bidentate nucleophile, condensing with the newly formed diketone to yield a hydrazone intermediate. This condensation is facilitated by the basic environment provided by pyridine and sodium dihydrogen phosphate, which helps to neutralize the hydrogen iodide byproduct and drive the equilibrium forward. The final cyclization step involves an intramolecular nucleophilic attack where the terminal nitrogen of the hydrazone attacks the remaining carbonyl carbon, closing the five-membered triazole ring. The presence of iodine throughout the process likely serves to regenerate the active oxidizing species and promote the dehydration steps necessary for aromatization of the triazole ring.
Impurity control in this system is inherently managed by the specificity of the iodine-mediated oxidation and the thermodynamic stability of the final triazole product. Unlike radical-based halogenations that can lead to poly-halogenated byproducts, the Kornblum oxidation is relatively selective for the alpha-position of the ketone. Furthermore, the use of stoichiometric amounts of sodium dihydrogen phosphate acts as a buffer, preventing the accumulation of acidic species that could catalyze the decomposition of the sensitive hydrazone intermediate or promote side reactions such as hydrolysis. The reaction conditions, specifically the temperature range of 110°C to 130°C, are optimized to ensure complete conversion of the intermediate while minimizing thermal degradation of the trifluoromethyl group. This precise control over the reaction environment ensures that the crude product profile is clean, significantly reducing the burden on downstream purification processes like column chromatography or recrystallization, which is vital for maintaining high purity standards required in pharmaceutical manufacturing.
How to Synthesize 3,4,5-Trisubstituted 1,2,4-Triazole Efficiently
The execution of this synthesis protocol requires careful attention to the sequential addition of reagents and temperature control to maximize yield and minimize byproduct formation. The process begins with the activation of the ketone substrate, followed by the introduction of the hydrazine component to trigger the cyclization cascade. Detailed operational parameters, including specific molar ratios and heating profiles, are critical for reproducibility and have been optimized through extensive experimentation as documented in the patent examples. For process chemists looking to implement this route, understanding the kinetics of the initial oxidation step versus the cyclization step is key to troubleshooting any potential stalls in conversion. The following guide outlines the standardized procedure derived from the patent data to ensure successful replication of this high-value transformation.
- Mix aryl ethyl ketone and elemental iodine in DMSO, heating to 90-110°C for 4-6 hours to initiate Kornblum oxidation.
- Add additional iodine, sodium dihydrogen phosphate, pyridine, and trifluoroethylimide hydrazide to the reaction mixture.
- Heat the solution to 110-130°C for 12-20 hours to complete the cyclization, followed by filtration and column chromatography purification.
Commercial Advantages for Procurement and Supply Chain Teams
From a strategic procurement perspective, this iodine-promoted methodology offers substantial advantages by fundamentally altering the cost drivers associated with heterocycle synthesis. The elimination of precious metal catalysts removes a significant variable cost component and mitigates the supply chain risks associated with the volatility of metal prices and availability. Moreover, the avoidance of heavy metals simplifies the purification train, potentially allowing for simpler crystallization processes rather than expensive chromatographic separations or scavenging treatments, which translates directly into reduced processing time and lower solvent consumption. The use of DMSO, a high-boiling polar aprotic solvent, allows for high-temperature reactions that accelerate kinetics, thereby increasing throughput capacity in existing reactor infrastructure without the need for cryogenic cooling or specialized high-pressure equipment. These factors collectively contribute to a more resilient and cost-effective supply chain for critical pharmaceutical intermediates.
- Cost Reduction in Manufacturing: The substitution of expensive transition metal catalysts with inexpensive elemental iodine represents a direct reduction in raw material costs, while the simplified workup procedures reduce labor and utility expenses associated with extended purification steps. The ability to run the reaction without strict anhydrous conditions further lowers costs by reducing the need for solvent drying and inert gas blanketing, making the process economically viable for large-scale production.
- Enhanced Supply Chain Reliability: The starting materials, specifically aryl ethyl ketones and trifluoroethylimide hydrazides, are commodity chemicals with established global supply chains, ensuring consistent availability and reducing the risk of production delays due to raw material shortages. The robustness of the reaction conditions means that the process is less sensitive to minor variations in reagent quality or environmental factors, leading to more predictable batch outcomes and reliable delivery schedules for downstream API manufacturers.
- Scalability and Environmental Compliance: The absence of toxic heavy metals significantly reduces the environmental footprint of the manufacturing process, easing the burden of wastewater treatment and hazardous waste disposal compliance. The process has been demonstrated to scale effectively from gram to multi-gram levels with consistent yields, indicating a clear path towards kilogram and tonne-scale production without the need for extensive process re-engineering, thus supporting rapid commercialization timelines.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These answers are derived directly from the experimental data and technical specifications provided in the patent documentation, offering clarity on the practical aspects of adopting this method for industrial applications. Understanding these nuances is essential for project managers and technical leads evaluating the feasibility of integrating this route into their existing manufacturing portfolios.
Q: Does this synthesis method require expensive heavy metal catalysts?
A: No, the process described in patent CN113105402B utilizes elemental iodine as a non-metal promoter, eliminating the need for costly and toxic transition metal catalysts typically found in traditional triazole synthesis.
Q: What are the optimal reaction conditions for scaling this process?
A: The reaction operates effectively in dimethyl sulfoxide (DMSO) at temperatures between 110°C and 130°C for the cyclization step, without requiring strict anhydrous or oxygen-free environments, which simplifies scale-up.
Q: Can this method accommodate diverse functional groups on the aromatic rings?
A: Yes, the method demonstrates broad substrate tolerance, successfully synthesizing derivatives with substituents such as methyl, methoxy, chloro, trifluoromethyl, and bromo groups on both the N-aryl and acyl moieties.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3,4,5-Trisubstituted 1,2,4-Triazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that advanced synthetic methodologies play in accelerating drug development and optimizing commercial production. Our team of expert process chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that innovative laboratory discoveries like the iodine-promoted triazole synthesis are seamlessly translated into robust industrial processes. We are committed to delivering high-purity pharmaceutical intermediates that meet stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation to verify identity and assay. Our dedication to quality and efficiency makes us an ideal partner for organizations seeking to secure a stable supply of complex heterocyclic building blocks.
We invite you to collaborate with us to explore how this cost-effective synthesis route can enhance your project economics. Please contact our technical procurement team to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and comprehensive route feasibility assessments to support your decision-making process and help you achieve your supply chain objectives with confidence.