Scalable Synthesis of Trifluoromethylated 1,2,4-Triazoles: A Metal-Free Route for Advanced Pharmaceutical Intermediates
Scalable Synthesis of Trifluoromethylated 1,2,4-Triazoles: A Metal-Free Route for Advanced Pharmaceutical Intermediates
In the rapidly evolving landscape of heterocyclic chemistry, the development of efficient synthetic routes for bioactive scaffolds remains a paramount challenge for research and development teams globally. Patent CN113105402B introduces a pivotal advancement in the preparation of 3,4,5-trisubstituted 1,2,4-triazole compounds, a structural motif ubiquitous in modern medicinal chemistry. These nitrogen-containing five-membered heterocycles serve as critical building blocks for high-value active pharmaceutical ingredients (APIs), including notable drugs such as Maraviroc, Sitagliptin, and Deferasirox, as illustrated in the structural diversity shown below. The introduction of a trifluoromethyl group into these heterocyclic systems is particularly strategic, as it significantly enhances physicochemical properties such as metabolic stability, lipophilicity, and bioavailability, thereby optimizing the pharmacokinetic profile of the final drug candidate.
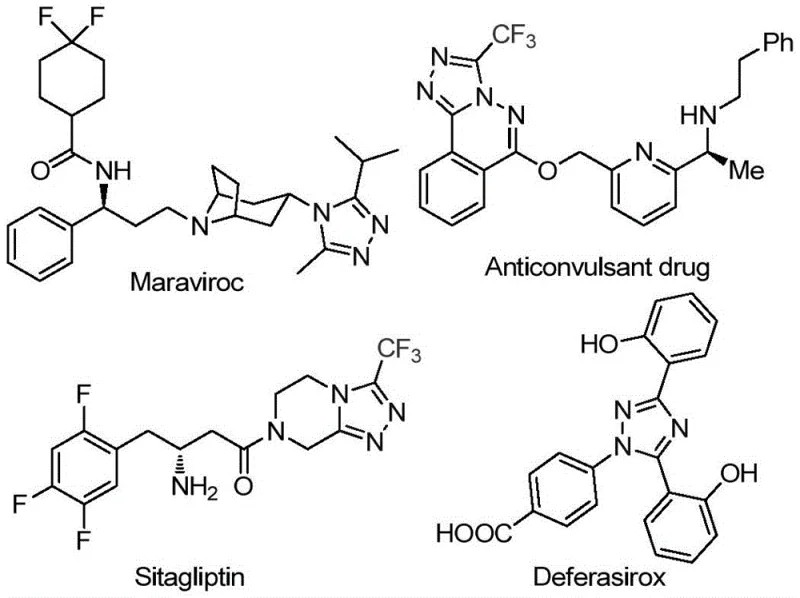
The significance of this patented methodology extends beyond mere academic interest; it addresses critical bottlenecks in the supply chain for reliable pharmaceutical intermediate suppliers. Traditional methods for constructing the 1,2,4-triazole core often rely on harsh conditions or expensive reagents that complicate downstream processing. By leveraging a non-metal promoted synthesis using readily available aryl ketones and trifluoroethylimide hydrazides, this invention offers a streamlined pathway that aligns perfectly with the industry's demand for greener, more cost-effective manufacturing processes. For procurement managers and supply chain heads, the implication is clear: a robust, scalable technology that reduces dependency on critical raw materials while maintaining high purity standards essential for regulatory compliance in drug manufacturing.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of polysubstituted 1,2,4-triazoles, particularly those bearing both trifluoromethyl and acyl groups, has been fraught with synthetic challenges that hinder large-scale production. Conventional protocols frequently necessitate the use of transition metal catalysts, which not only inflate the raw material costs but also introduce significant complications regarding residual metal removal—a critical quality attribute for any pharmaceutical intermediate intended for human use. Furthermore, many existing literature methods require stringent reaction conditions, such as strictly anhydrous and oxygen-free environments, which demand specialized equipment like Schlenk lines or gloveboxes. These operational constraints drastically increase capital expenditure (CAPEX) and operational expenditure (OPEX), rendering such processes economically unviable for commercial scale-up of complex pharmaceutical intermediates. Additionally, the limited substrate scope of older methods often fails to accommodate diverse functional groups, restricting the chemical space available for medicinal chemists during lead optimization phases.
The Novel Approach
In stark contrast to these legacy techniques, the novel approach detailed in the patent utilizes a simple, efficient, and metal-free strategy promoted by elemental iodine. This method capitalizes on the reactivity of aryl ethyl ketones, which undergo an iodination and subsequent Kornblum oxidation in dimethyl sulfoxide (DMSO) to generate reactive aryl diketone intermediates in situ. These intermediates then engage in a tandem cyclization with trifluoroethylimide hydrazides to forge the desired triazole ring system. The elegance of this route lies in its operational simplicity; it proceeds smoothly in standard organic solvents without the need for inert atmospheres, thereby democratizing access to these valuable scaffolds. As depicted in the general reaction scheme below, the transformation is direct and high-yielding, utilizing cheap and commercially available starting materials that ensure a stable and continuous supply chain for manufacturing facilities.
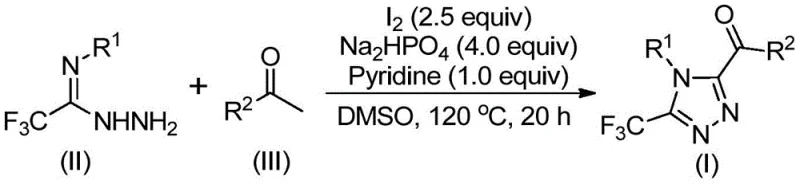
Mechanistic Insights into Iodine-Promoted Tandem Cyclization
To fully appreciate the technical robustness of this synthesis, one must delve into the mechanistic intricacies that drive the formation of the 3,4,5-trisubstituted 1,2,4-triazole core. The reaction initiates with the activation of the aryl ethyl ketone by elemental iodine in DMSO, facilitating an alpha-iodination followed by a Kornblum oxidation. This sequence effectively converts the methyl ketone into a highly electrophilic 1,2-dicarbonyl species, which serves as the crucial linchpin for the subsequent heterocycle formation. The presence of DMSO is not merely as a solvent but acts as an oxygen donor in the oxidation step, a dual role that streamlines the reagent list and minimizes waste generation. This initial activation is critical because it generates the necessary electronic bias on the carbon backbone to accept the nucleophilic attack from the hydrazide nitrogen.
Following the generation of the dicarbonyl intermediate, the trifluoroethylimide hydrazide enters the cycle, undergoing a dehydration condensation to form a hydrazone intermediate. This step is facilitated by the basic environment provided by pyridine and sodium dihydrogen phosphate, which help to neutralize the hydrogen iodide byproduct and drive the equilibrium forward. The final and most decisive step is the intramolecular cyclization, promoted by the continued presence of iodine and base. This cyclization closes the five-membered ring, establishing the 1,2,4-triazole architecture with the trifluoromethyl group securely positioned at the 3-position and the acyl group at the 5-position. The mechanism ensures high regioselectivity and minimizes the formation of isomeric impurities, a key factor for R&D directors focused on purity and impurity profiles. The broad substrate tolerance, allowing for various substituents like methoxy, chloro, and alkyl groups as shown in the specific examples below, underscores the versatility of this mechanistic pathway.
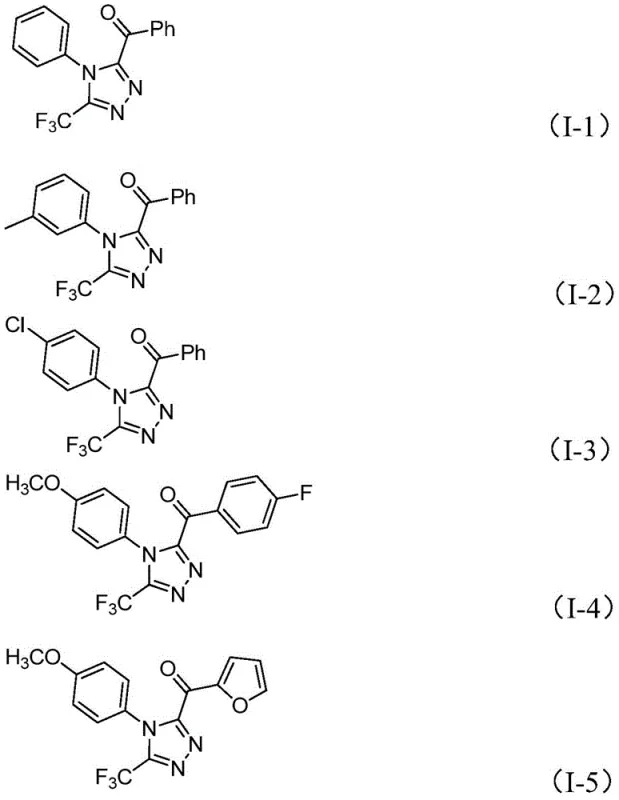
How to Synthesize 3,4,5-Trisubstituted 1,2,4-Triazole Efficiently
The practical execution of this synthesis is designed for ease of operation, making it accessible for both laboratory discovery and pilot plant operations. The protocol involves a sequential addition of reagents where temperature control is the primary variable, eliminating the need for complex pressure vessels or cryogenic cooling. By adhering to the optimized molar ratios and thermal profiles defined in the patent, manufacturers can achieve consistent results across different batches. The detailed standardized synthesis steps, including precise stoichiometry and workup procedures, are outlined in the guide below to ensure reproducibility and safety during implementation.
- Mix aryl ethyl ketone and elemental iodine in DMSO solvent and heat to 90-110°C for 4-6 hours to initiate oxidation.
- Add trifluoroethylimide hydrazide, sodium dihydrogen phosphate, pyridine, and additional iodine to the reaction mixture.
- Heat the mixture to 110-130°C for 12-20 hours to complete the cyclization, followed by filtration and column chromatography purification.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain leaders, the adoption of this iodine-promoted methodology represents a strategic opportunity to optimize the cost structure of API manufacturing while enhancing supply security. The elimination of precious metal catalysts removes a significant volatility factor from the raw material basket, as the prices of metals like palladium or copper can fluctuate wildly based on geopolitical factors. Instead, the process relies on elemental iodine and DMSO, commodities that are produced in massive volumes globally, ensuring price stability and ready availability. Furthermore, the avoidance of heavy metals simplifies the downstream purification process, as there is no need for expensive scavenging resins or complex extraction protocols to meet strict residual metal limits imposed by regulatory bodies like the FDA or EMA.
- Cost Reduction in Manufacturing: The economic benefits of this process are driven primarily by the simplification of the reaction setup and the reduction in reagent costs. By operating without the need for anhydrous or oxygen-free conditions, facilities can utilize standard stainless steel reactors rather than specialized glass-lined or Hastelloy vessels required for sensitive organometallic chemistry. This reduction in equipment specification translates directly to lower capital depreciation costs per kilogram of product. Additionally, the high atom economy and the use of inexpensive promoters mean that the cost of goods sold (COGS) for these intermediates can be significantly lowered, providing a competitive edge in tender negotiations for generic drug formulations.
- Enhanced Supply Chain Reliability: Supply chain resilience is bolstered by the use of robust, commodity-grade chemicals that are less susceptible to logistics disruptions. Unlike specialized ligands or catalysts that may have single-source suppliers with long lead times, iodine and aryl ketones are available from a wide network of global chemical distributors. This multi-sourcing capability mitigates the risk of production stoppages due to raw material shortages. Moreover, the scalability of the reaction from gram to kilogram levels without loss of efficiency ensures that the transition from clinical trial material to commercial launch stock can be executed seamlessly, reducing the lead time for high-purity pharmaceutical intermediates.
- Scalability and Environmental Compliance: From an environmental, health, and safety (EHS) perspective, this method offers substantial advantages that align with modern green chemistry principles. The absence of toxic heavy metals reduces the burden on wastewater treatment facilities and lowers the cost of hazardous waste disposal. The reaction conditions are moderate, avoiding extreme pressures or temperatures that pose safety risks in large-scale plants. This inherent safety profile facilitates easier permitting and regulatory approval for new manufacturing lines. Consequently, the process supports sustainable manufacturing goals, allowing companies to reduce their environmental footprint while maintaining high throughput and operational efficiency in the production of complex fine chemicals.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and beneficial effects reported in the patent documentation, providing clarity on the feasibility and advantages of adopting this route for your specific project needs. Understanding these details is crucial for making informed decisions about process integration and vendor selection.
Q: Does this synthesis method require expensive transition metal catalysts?
A: No, the method described in patent CN113105402B utilizes elemental iodine as a promoter, completely eliminating the need for costly and toxic heavy metal catalysts typically used in triazole synthesis.
Q: What are the optimal reaction conditions for scaling this process?
A: The process operates effectively in DMSO at temperatures between 110-130°C without requiring strict anhydrous or oxygen-free conditions, making it highly suitable for industrial scale-up.
Q: Can this method accommodate diverse functional groups on the aryl rings?
A: Yes, the protocol demonstrates excellent functional group tolerance, successfully synthesizing derivatives with methyl, methoxy, chloro, and trifluoromethyl substituents on both aromatic rings.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3,4,5-Trisubstituted 1,2,4-Triazole Supplier
As the global demand for fluorinated heterocycles continues to surge, partnering with a technically proficient CDMO is essential for navigating the complexities of modern drug development. NINGBO INNO PHARMCHEM possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project can transition smoothly from benchtop discovery to full-scale manufacturing. Our commitment to quality is underpinned by stringent purity specifications and rigorous QC labs equipped with state-of-the-art analytical instrumentation, guaranteeing that every batch of 3,4,5-trisubstituted 1,2,4-triazole meets the highest international standards for pharmaceutical applications.
We invite you to leverage our expertise to optimize your supply chain and accelerate your time-to-market. By engaging with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements and purity needs. We encourage potential partners to reach out for specific COA data and route feasibility assessments to determine how this innovative iodine-promoted synthesis can be integrated into your existing portfolio, delivering both technical excellence and commercial value.