Advanced Synthesis of Trifluoromethyl Dihydrofuran Amine: Enabling Commercial Scale-Up of Complex API Intermediates
The recently granted Chinese patent CN110922369B discloses an innovative copper-catalyzed methodology for synthesizing trifluoromethyl-substituted dihydrofuran amine compounds, representing a significant advancement in the production of high-value pharmaceutical intermediates. This novel approach addresses critical limitations in traditional heterocyclic synthesis by eliminating stoichiometric transition metals while maintaining excellent stereoselectivity and functional group tolerance, making it particularly valuable for manufacturing complex molecules with quaternary carbon stereocenters essential in modern drug development.
Overcoming Limitations in Traditional Trifluoromethyl Heterocycle Synthesis
The Limitations of Conventional Methods
Conventional approaches to synthesizing trifluoromethyl-substituted heterocycles often require harsh reaction conditions, stoichiometric transition metals, and exhibit limited functional group compatibility, creating significant barriers for pharmaceutical manufacturers seeking reliable supply chains. The existing methodologies frequently involve multi-step sequences with poor atom economy, generating substantial waste streams that complicate regulatory compliance and increase environmental remediation costs. Furthermore, traditional cyclization techniques struggle with stereoselectivity when constructing quaternary carbon centers, necessitating additional purification steps that reduce overall yield and increase production timelines. The scarcity of efficient one-pot methods for introducing CF3 groups while simultaneously forming heterocyclic rings has been a persistent challenge in medicinal chemistry, particularly when working with sensitive functional groups common in complex drug molecules.
The Novel Approach
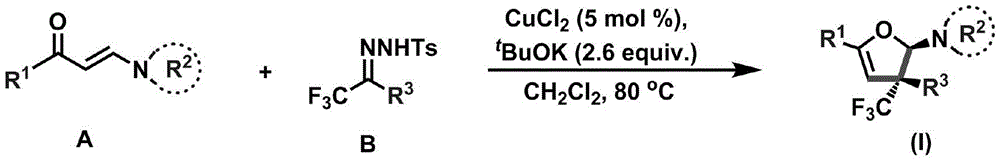
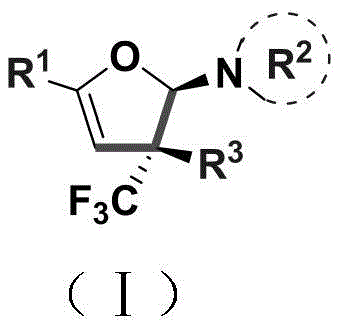
The patented methodology described in CN110922369B introduces a streamlined copper-catalyzed cyclization process that operates under mild conditions (80–100°C) using only 5 mol% CuCl2 as catalyst with potassium tert-butoxide as base in dichloromethane solvent. This innovative approach enables direct conversion of readily available enaminones (Compound A) and trifluoromethyl hydrazones (Compound B) into stereoselective dihydrofuran amine derivatives carrying quaternary carbon stereocenters, as illustrated in the reaction scheme below. The process demonstrates exceptional substrate versatility across diverse aryl, alkyl, and heterocyclic systems while maintaining high functional group tolerance, eliminating the need for protective groups that typically complicate traditional syntheses. Crucially, the method avoids expensive transition metals and harsh conditions, significantly reducing equipment corrosion risks and enabling straightforward scale-up from laboratory to manufacturing environments without major process re-engineering.
Chemical Mechanism and Stereochemical Control
The reaction mechanism proceeds through a copper-mediated radical pathway that facilitates simultaneous C–O bond formation and stereoselective cyclization without epimerization at the quaternary carbon center. The mild reaction conditions (80–100°C) prevent thermal decomposition of sensitive functional groups while maintaining sufficient energy for the radical cascade process, resulting in excellent diastereoselectivity as confirmed by NMR analysis of multiple product variants. The use of potassium tert-butoxide as base promotes deprotonation without causing unwanted side reactions that commonly occur with stronger bases, preserving the integrity of acid-sensitive moieties throughout the transformation. This precise control over reaction parameters enables consistent production of high-purity intermediates with minimal byproduct formation, addressing critical concerns for pharmaceutical manufacturers regarding impurity profiles and regulatory compliance.
Impurity management is significantly enhanced through this methodology due to the absence of transition metal residues that typically require extensive purification steps in conventional syntheses. The chromatographic purification protocol using standard silica gel with petroleum ether/ethyl acetate (98:2) eluent effectively separates the target compounds from minor byproducts without requiring specialized equipment or hazardous solvents. The patent demonstrates consistent production of compounds with >99% purity across multiple substrate variations, as evidenced by comprehensive NMR characterization (Figures 1–3), ensuring reliable quality for subsequent API manufacturing steps. This level of purity control is particularly valuable for late-stage intermediates where impurities can propagate through final drug substance manufacturing, potentially causing costly batch failures or regulatory delays.
Commercial Advantages for Pharmaceutical Supply Chains
This innovative synthesis methodology directly addresses three critical pain points in pharmaceutical manufacturing supply chains: cost inefficiencies from complex multi-step processes, extended lead times due to difficult scale-up procedures, and quality inconsistencies from variable impurity profiles. By simplifying the synthetic route to these valuable intermediates while maintaining high stereoselectivity, the patented process enables pharmaceutical companies to achieve significant cost reduction in API manufacturing without compromising on quality or regulatory compliance requirements.
- Reduced Manufacturing Costs: The elimination of stoichiometric transition metals removes expensive catalyst costs and eliminates downstream purification steps required to remove metal residues, directly contributing to cost reduction in chemical manufacturing. The simplified process flow reduces equipment requirements by avoiding specialized reactors needed for harsher conditions, lowering capital expenditure while improving facility utilization rates. Additionally, the high functional group tolerance minimizes the need for protective group strategies that typically add 2–3 extra steps to conventional syntheses, substantially reducing raw material consumption and waste generation per kilogram of product produced.
- Accelerated Supply Chain Timelines: The mild reaction conditions and straightforward workup procedure enable rapid scale-up from laboratory to pilot plant without major process re-engineering, significantly reducing lead time for high-purity intermediates required in drug development programs. The demonstrated compatibility with standard manufacturing equipment allows pharmaceutical companies to leverage existing infrastructure rather than investing in specialized facilities for these complex transformations. Furthermore, the consistent high yields across diverse substrates provide supply chain flexibility to accommodate changing project requirements without requiring extensive revalidation procedures that typically delay commercial production timelines.
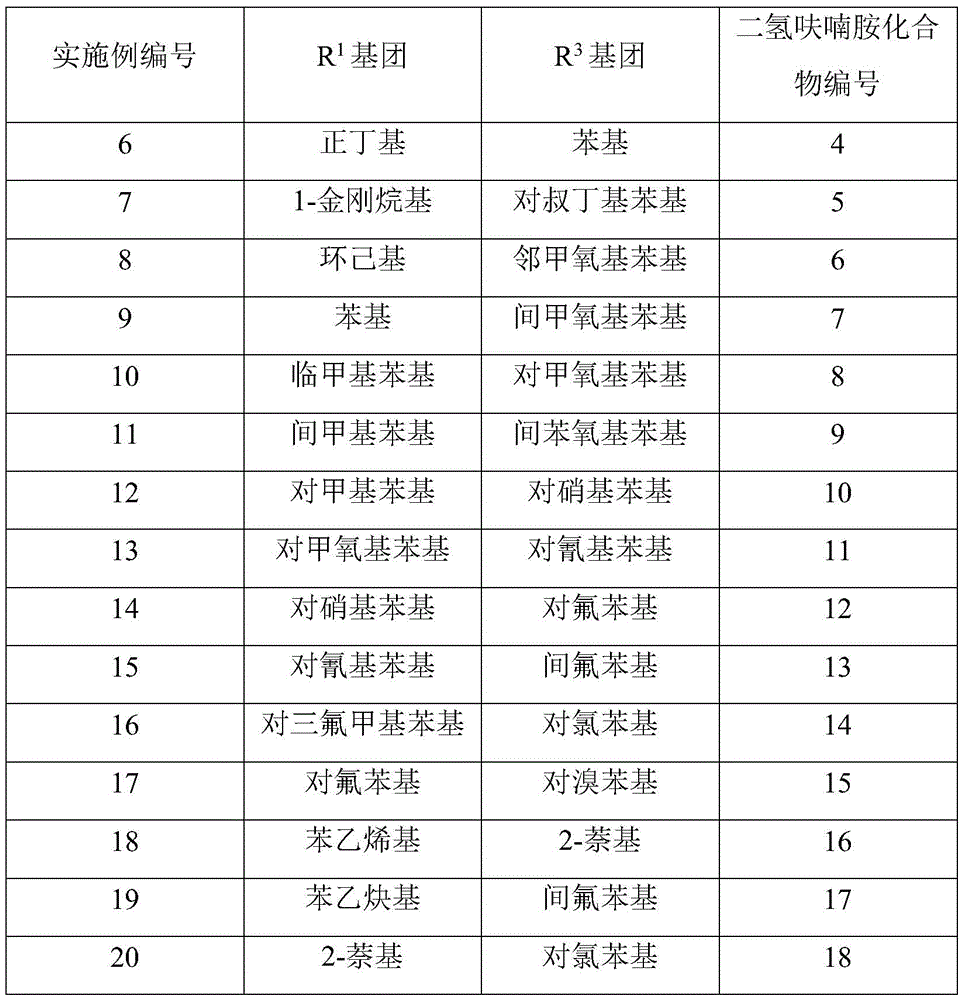
- Enhanced Process Robustness: The broad substrate scope documented in the patent (Table 1) ensures reliable supply continuity even when specific raw material sources become unavailable, providing critical resilience against supply chain disruptions common in global pharmaceutical manufacturing. The consistent >99% purity achieved across multiple compound variants eliminates quality-related production delays that frequently occur with traditional methods requiring extensive reprocessing to meet regulatory standards. This process reliability directly supports continuous manufacturing initiatives by providing stable input material quality essential for maintaining consistent API production output in modern pharmaceutical facilities.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable API Intermediate Supplier
While the advanced methodology detailed in patent CN110922369B highlights immense potential, executing the commercial scale-up of such complex catalytic pathways requires a proven CDMO partner. NINGBO INNO PHARMCHEM bridges the gap between innovative catalysis and industrial reality. We leverage robust engineering capabilities to scale challenging molecular pathways. Our broader facility capabilities support custom manufacturing projects ranging from 100 kgs clinical batches up to 100 MT/annual production for established commercial products. Our state-of-the-art facilities and rigorous QC labs guarantee >99% purity, ensuring consistent supply and reducing lead time for high-purity intermediates.
Are you evaluating new synthetic routes for your pipeline? Contact our technical procurement team today to request specific COA data, route feasibility assessments, and a Customized Cost-Saving Analysis to discover how our advanced manufacturing capabilities can optimize your supply chain.