Advanced Metal-Free Synthesis of 3,4,5-Trisubstituted 1,2,4-Triazoles for Pharmaceutical Applications
Advanced Metal-Free Synthesis of 3,4,5-Trisubstituted 1,2,4-Triazoles for Pharmaceutical Applications
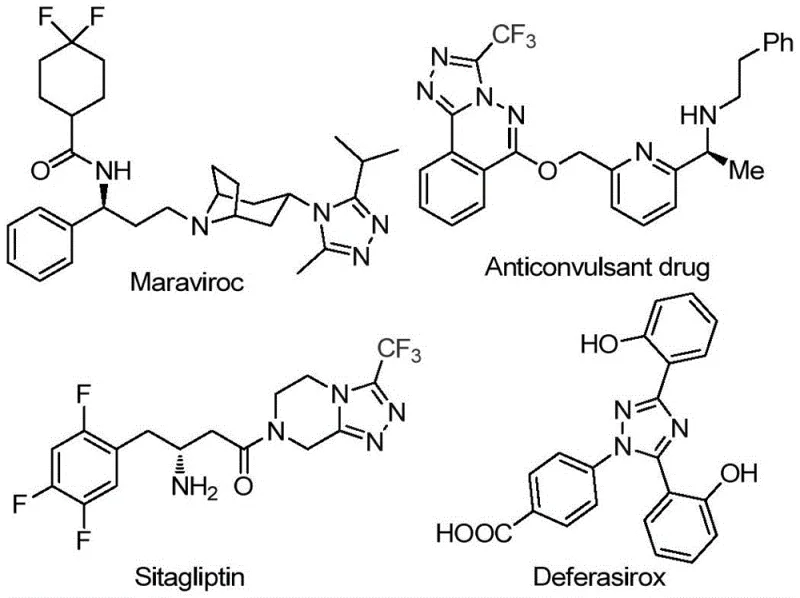
The pharmaceutical industry continuously seeks robust and scalable synthetic routes for nitrogen-containing heterocycles, particularly 1,2,4-triazoles, which serve as critical scaffolds in numerous bioactive molecules. Patent CN113105402B discloses a groundbreaking preparation method for 3,4,5-trisubstituted 1,2,4-triazole compounds that addresses long-standing challenges in efficiency and environmental compliance. This technology leverages a non-metallic iodine-promoted cascade reaction, eliminating the dependency on scarce transition metals while maintaining high yields and purity. The significance of this advancement cannot be overstated, as triazole motifs are ubiquitous in high-value drugs such as Maraviroc, Sitagliptin, and Deferasirox, where the introduction of trifluoromethyl groups enhances metabolic stability and lipophilicity. By utilizing cheap and readily available starting materials like aryl ethyl ketones and trifluoroethylimide hydrazides, this process offers a compelling value proposition for manufacturers aiming to optimize their supply chains for complex pharmaceutical intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of polysubstituted 1,2,4-triazoles has relied heavily on transition metal catalysis or harsh reaction conditions that pose significant operational risks and cost burdens. Traditional pathways often require stringent anhydrous and oxygen-free environments, necessitating specialized equipment and increasing energy consumption substantially. Furthermore, the use of heavy metal catalysts introduces complex downstream processing requirements, as residual metal levels must be reduced to parts-per-million specifications to meet regulatory standards for active pharmaceutical ingredients (APIs). These purification steps not only extend production lead times but also generate substantial hazardous waste, conflicting with modern green chemistry principles. Additionally, many existing methods suffer from limited substrate scope, failing to tolerate sensitive functional groups or struggling to introduce specific substituents like trifluoromethyl and acyl groups simultaneously, thereby restricting their utility in diverse drug discovery programs.
The Novel Approach
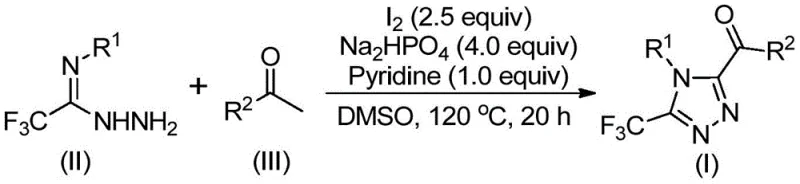
In stark contrast, the methodology outlined in Patent CN113105402B presents a streamlined, metal-free alternative that fundamentally reshapes the production landscape for these valuable heterocycles. The core innovation lies in the use of elemental iodine and dimethyl sulfoxide (DMSO) to facilitate a tandem iodination and Kornblum oxidation sequence, converting simple aryl ketones into reactive intermediates in situ. This approach operates under relatively mild thermal conditions without the need for inert atmosphere protection, drastically simplifying reactor setup and operation. The reaction proceeds through a cascade mechanism where the generated aryl diketones condense with trifluoroethylimide hydrazides to form the final triazole ring system efficiently. As illustrated in the reaction scheme below, this one-pot strategy minimizes isolation steps, reduces solvent usage, and significantly lowers the overall cost of goods sold (COGS) by utilizing commodity chemicals as feedstocks.
Mechanistic Insights into Iodine-Promoted Cyclization
A deep understanding of the reaction mechanism is crucial for R&D directors aiming to implement this technology at scale. The process initiates with the iodination of the aryl ethyl ketone, followed by a Kornblum oxidation mediated by DMSO to generate an α-dicarbonyl species. This oxidative transformation is pivotal, as it activates the methyl group adjacent to the carbonyl, rendering it susceptible to nucleophilic attack. Subsequently, the trifluoroethylimide hydrazide acts as a bidentate nucleophile, undergoing dehydration condensation with the dicarbonyl intermediate to form a hydrazone species. The final cyclization step is promoted by the synergistic action of iodine and the base system (sodium dihydrogen phosphate and pyridine), which facilitates intramolecular ring closure to yield the stable 1,2,4-triazole core. This mechanistic pathway ensures high atom economy and minimizes the formation of polymeric byproducts often seen in radical-based halogenation reactions.
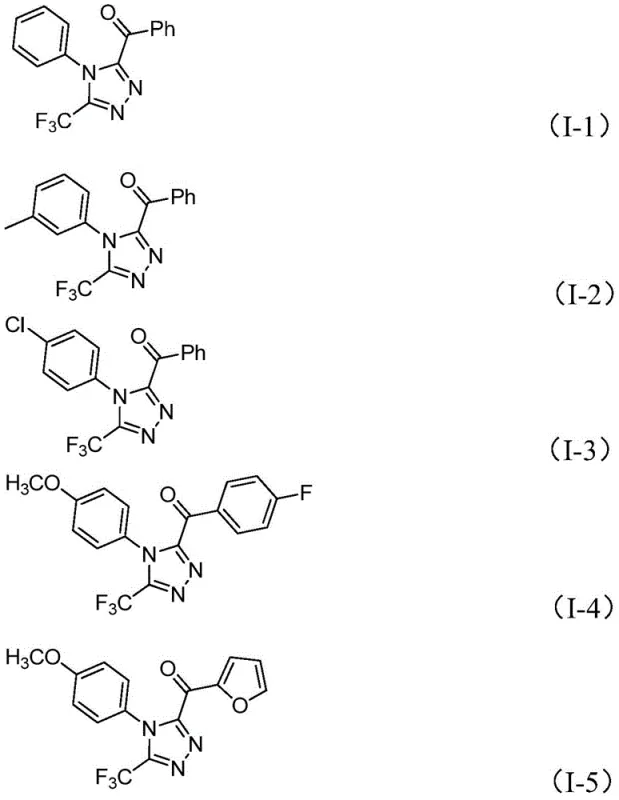
From an impurity control perspective, the choice of reagents plays a vital role in ensuring product quality. The use of sodium dihydrogen phosphate provides a buffered environment that prevents the degradation of acid-sensitive functional groups, while pyridine acts as both a solvent co-component and a base to neutralize generated hydrogen iodide. The specific molar ratios defined in the patent, such as the 4:1:2.5 ratio of phosphate to pyridine to iodine, are optimized to drive the equilibrium towards the desired triazole product while suppressing side reactions like over-iodination or hydrolysis. The broad substrate tolerance allows for the incorporation of various electron-donating and electron-withdrawing groups, as evidenced by the successful synthesis of derivatives containing methoxy, chloro, and trifluoromethyl substituents, ensuring that the process is robust enough for the synthesis of diverse pharmaceutical building blocks.
How to Synthesize 3,4,5-Trisubstituted 1,2,4-Triazoles Efficiently
Implementing this synthesis route requires precise control over reaction parameters to maximize yield and purity. The protocol involves a two-stage heating process where the initial oxidation is conducted at lower temperatures before ramping up for the cyclization phase. Operators must ensure thorough mixing during the addition of solid reagents to prevent localized hot spots that could lead to decomposition. The post-treatment process is notably straightforward, involving simple filtration followed by silica gel chromatography, which is a standard unit operation in most fine chemical facilities. For detailed operational parameters and safety guidelines, please refer to the standardized synthesis steps provided below.
- Mix aryl ethyl ketone and iodine in DMSO, heating to 90-110°C for 4-6 hours to initiate Kornblum oxidation.
- Add additional iodine, sodium dihydrogen phosphate, pyridine, and trifluoroethylimide hydrazide to the reaction mixture.
- Heat the mixture to 110-130°C for 12-20 hours, then filter and purify via column chromatography to isolate the product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this metal-free synthesis route offers transformative benefits regarding cost stability and logistical reliability. By eliminating the need for precious metal catalysts such as palladium or copper, manufacturers can decouple their production costs from the volatile commodities market associated with these metals. Furthermore, the removal of heavy metal catalysts obviates the need for expensive scavenging resins or complex extraction protocols typically required to meet strict residual metal specifications, resulting in significant operational expenditure savings. The use of DMSO as a primary solvent is another strategic advantage, as it is a high-boiling, polar aprotic solvent that is widely available globally at competitive prices, ensuring supply continuity even during market fluctuations.
- Cost Reduction in Manufacturing: The economic viability of this process is driven by the utilization of commodity starting materials like acetophenones and hydrazides, which are produced on a multi-ton scale globally. The absence of expensive ligands or specialized catalysts drastically reduces the raw material bill. Moreover, the simplified workup procedure reduces labor hours and solvent consumption during purification, contributing to a leaner manufacturing cost structure. The ability to run the reaction without rigorous exclusion of moisture or oxygen further lowers infrastructure costs, as standard glass-lined or stainless steel reactors can be utilized without modification.
- Enhanced Supply Chain Reliability: Sourcing risks are minimized because all key reagents, including elemental iodine and sodium dihydrogen phosphate, are bulk chemicals with established global supply chains. Unlike proprietary catalysts that may be sourced from single vendors, these materials are available from multiple suppliers, mitigating the risk of supply disruption. The robustness of the reaction conditions means that production can be easily transferred between different manufacturing sites or contract manufacturing organizations (CMOs) without extensive re-validation, providing flexibility in capacity planning and inventory management.
- Scalability and Environmental Compliance: The process is inherently scalable, having been demonstrated to work efficiently from gram to multi-kilogram scales with consistent results. The avoidance of toxic heavy metals aligns with increasingly stringent environmental regulations regarding wastewater discharge and solid waste disposal. This green chemistry profile not only reduces the environmental footprint of the manufacturing process but also simplifies the regulatory filing process for new drug applications, as the impurity profile is cleaner and easier to characterize compared to metal-catalyzed alternatives.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this triazole synthesis technology. These insights are derived directly from the experimental data and claims within Patent CN113105402B, providing a factual basis for decision-making. Understanding these nuances is essential for evaluating the feasibility of integrating this route into existing production portfolios.
Q: Does this synthesis require expensive heavy metal catalysts?
A: No, the method described in Patent CN113105402B utilizes elemental iodine as a promoter, completely avoiding the need for toxic or expensive transition metal catalysts, which simplifies purification and reduces environmental impact.
Q: What are the optimal reaction conditions for scaling this process?
A: The process operates effectively in DMSO solvent without strict anhydrous or oxygen-free conditions. The optimal temperature profile involves an initial stage at 90-110°C followed by a cyclization stage at 110-130°C, making it highly suitable for standard industrial reactors.
Q: Can this method accommodate diverse functional groups on the aromatic rings?
A: Yes, the methodology demonstrates excellent functional group tolerance, successfully synthesizing derivatives with methyl, methoxy, chloro, trifluoromethyl, and heteroaryl substituents, providing high versatility for medicinal chemistry applications.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3,4,5-Trisubstituted 1,2,4-Triazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in the success of your drug development programs. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet your volume requirements with consistency and precision. We are committed to delivering products that meet stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation. Our expertise in heterocyclic chemistry allows us to navigate complex synthetic challenges, ensuring that the transition from laboratory scale to industrial manufacturing is seamless and efficient.
We invite you to collaborate with us to leverage this advanced synthesis technology for your specific projects. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your volume needs, demonstrating how this metal-free route can optimize your budget. Please contact us to request specific COA data for our triazole intermediates and to discuss route feasibility assessments for your target molecules. Let us be your partner in driving innovation and efficiency in pharmaceutical manufacturing.