Scalable Metal-Free Synthesis of Trifluoromethyl-Triazoles for Advanced Pharmaceutical Intermediates
Scalable Metal-Free Synthesis of Trifluoromethyl-Triazoles for Advanced Pharmaceutical Intermediates
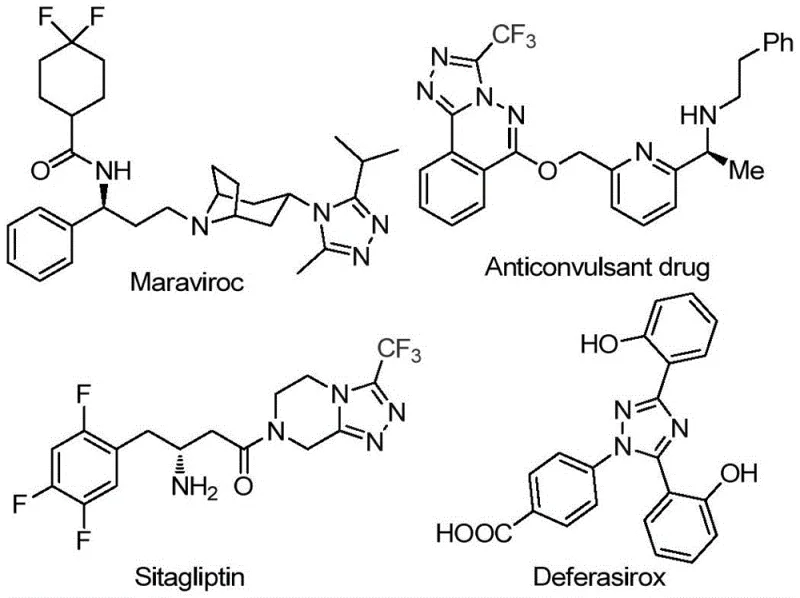
The rapid evolution of medicinal chemistry demands robust synthetic routes for nitrogen-containing heterocycles, particularly those incorporating fluorine motifs which enhance metabolic stability and bioavailability. Patent CN113105402B discloses a groundbreaking preparation method for 3,4,5-trisubstituted 1,2,4-triazole compounds, a scaffold prevalent in high-value therapeutics such as sitagliptin and maraviroc. This technology addresses critical bottlenecks in traditional heterocycle synthesis by replacing toxic heavy metal catalysts with an inexpensive iodine-promoted system. For R&D directors and procurement specialists seeking a reliable pharmaceutical intermediate supplier, this innovation represents a paradigm shift towards greener, more cost-effective manufacturing processes that do not compromise on purity or structural complexity.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of polysubstituted 1,2,4-triazole rings has relied heavily on transition metal catalysis or harsh cyclization conditions that pose significant challenges for industrial application. Traditional pathways often necessitate stringent anhydrous and oxygen-free environments, requiring specialized equipment and increasing operational expenditures substantially. Furthermore, the reliance on precious metal catalysts introduces complex downstream purification steps to remove trace metal impurities, which is a critical failure point for API manufacturing compliance. These legacy methods frequently suffer from limited substrate tolerance, failing to accommodate diverse functional groups without significant yield degradation, thereby restricting the chemical space available for drug discovery teams exploring novel analogs.
The Novel Approach
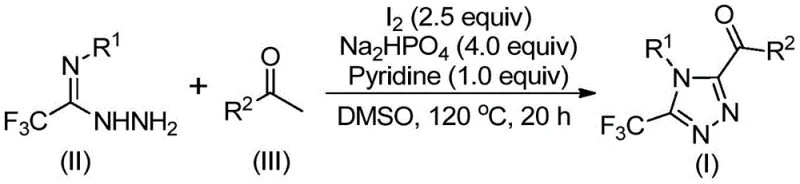
The methodology outlined in the patent introduces a streamlined, metal-free alternative that leverages elemental iodine and dimethyl sulfoxide (DMSO) to drive the reaction forward efficiently. By utilizing a tandem iodination and Kornblum oxidation sequence, the process converts readily available aryl ethyl ketones into reactive intermediates in situ, which subsequently undergo cyclization with trifluoroethylimide hydrazides. This approach eliminates the need for pre-functionalized starting materials and avoids the use of toxic heavy metals entirely. The reaction proceeds under relatively mild thermal conditions without the necessity for inert atmosphere protection, drastically simplifying the operational protocol and making it highly attractive for large-scale commercial production of complex pharmaceutical intermediates.
Mechanistic Insights into Iodine-Promoted Tandem Cyclization
The core of this synthetic breakthrough lies in the dual role of iodine as both an oxidant and a promoter within the DMSO solvent system. Initially, the aryl ethyl ketone undergoes iodination followed by Kornblum oxidation to generate an aryl diketone species. This highly reactive intermediate then condenses with the trifluoroethylimide hydrazide through a dehydration step to form a hydrazone intermediate. Subsequent intramolecular cyclization, facilitated by the basic environment provided by pyridine and sodium dihydrogen phosphate, closes the triazole ring. This mechanistic pathway ensures high atom economy and minimizes the formation of side products, resulting in a cleaner crude reaction profile that simplifies final purification efforts.
Impurity control is inherently managed by the selectivity of the iodine-mediated oxidation, which favors the formation of the desired 1,2,4-triazole core over potential isomeric byproducts. The use of sodium dihydrogen phosphate acts as a buffer to maintain optimal pH levels throughout the extended heating period, preventing the decomposition of sensitive trifluoromethyl groups. Experimental data indicates that the reaction tolerates a wide range of electronic effects on the aromatic rings, with electron-donating groups like methoxy and electron-withdrawing groups like chloro both yielding stable products. This robustness suggests that the mechanism is resilient against variations in substrate electronics, providing a versatile platform for synthesizing diverse libraries of trifluoromethylated heterocycles for biological screening.
How to Synthesize 3,4,5-Trisubstituted 1,2,4-Triazole Efficiently
The practical implementation of this synthesis involves a straightforward one-pot procedure that can be executed with standard laboratory glassware or adapted for reactor vessels. The process begins with the mixing of aryl ethyl ketone and iodine in DMSO, followed by a controlled heating phase to initiate oxidation. After the initial transformation, the hydrazide component and base additives are introduced directly into the same vessel, allowing the cyclization to proceed seamlessly. Detailed standardized synthesis steps see the guide below.
- Mix aryl ethyl ketone and iodine in DMSO, heating to 90-110°C for 4-6 hours to initiate oxidation.
- Add sodium dihydrogen phosphate, pyridine, and trifluoroethylimide hydrazide to the mixture.
- Heat the reaction to 110-130°C for 12-20 hours, then filter and purify via column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
From a supply chain perspective, this patented method offers transformative benefits by decoupling production from the volatility of precious metal markets and complex waste management protocols. The reliance on commodity chemicals such as elemental iodine, acetophenone derivatives, and DMSO ensures a stable and predictable raw material supply chain. For procurement managers, this translates to reduced exposure to price fluctuations associated with palladium or copper catalysts, while also lowering the total cost of ownership through simplified waste disposal procedures. The ability to run reactions without rigorous exclusion of air and moisture further reduces infrastructure costs, allowing for production in facilities that may not be equipped with specialized glovebox or Schlenk line capabilities.
- Cost Reduction in Manufacturing: The elimination of expensive transition metal catalysts removes a significant line item from the bill of materials, directly impacting the gross margin of the final intermediate. Additionally, the avoidance of heavy metals negates the need for costly scavenging resins or extensive chromatographic purification steps typically required to meet regulatory limits for residual metals in APIs. This streamlining of the downstream processing workflow results in substantial cost savings and shorter batch cycle times, enhancing overall plant throughput and profitability for contract manufacturing organizations.
- Enhanced Supply Chain Reliability: The starting materials, including various substituted acetophenones and trifluoroethylimide hydrazides, are commercially available in bulk quantities from multiple global suppliers. This diversity in sourcing options mitigates the risk of single-supplier dependency and ensures continuity of supply even during market disruptions. Furthermore, the robustness of the reaction conditions means that production schedules are less likely to be impacted by environmental factors or minor deviations in reagent quality, providing a more reliable delivery timeline for downstream pharmaceutical customers.
- Scalability and Environmental Compliance: The process demonstrates excellent scalability, having been validated from gram-scale experiments to potential multi-kilogram production runs without loss of efficiency. The use of DMSO, a high-boiling polar aprotic solvent, facilitates heat transfer and mixing in large reactors, while the absence of toxic heavy metals significantly reduces the environmental footprint of the manufacturing process. This alignment with green chemistry principles not only simplifies regulatory compliance regarding hazardous waste discharge but also enhances the sustainability profile of the final product, a key metric for modern pharmaceutical supply chains.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the scalability, safety, and applicability of this iodine-promoted synthesis route. These insights are derived directly from the experimental data and scope limitations defined within the patent documentation, providing clarity for technical teams evaluating this technology for adoption.
Q: Does this synthesis require expensive transition metal catalysts?
A: No, the patented method utilizes elemental iodine as a non-metallic promoter, eliminating the need for costly heavy metal catalysts and simplifying purification.
Q: What are the optimal reaction conditions for scale-up?
A: The process operates effectively in DMSO at temperatures between 110-130°C without strict anhydrous or oxygen-free conditions, facilitating easier industrial scale-up.
Q: What is the substrate scope for R1 and R2 groups?
A: The method tolerates various substituents including methyl, methoxy, chloro, and trifluoromethyl groups on both aryl and heteroaryl rings, yielding products with 37-86% efficiency.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3,4,5-Trisubstituted 1,2,4-Triazole Supplier
As the demand for fluorinated heterocycles continues to surge in the development of next-generation therapeutics, partnering with an experienced CDMO is essential for navigating the complexities of process scale-up. NINGBO INNO PHARMCHEM possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project transitions smoothly from benchtop discovery to full-scale manufacturing. Our facility is equipped with stringent purity specifications and rigorous QC labs capable of analyzing complex impurity profiles, guaranteeing that every batch of 3,4,5-trisubstituted 1,2,4-triazole meets the highest international standards for pharmaceutical intermediates.
We invite you to leverage our technical expertise to optimize your supply chain and reduce time-to-market for your critical drug candidates. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and route feasibility assessments to demonstrate how our advanced synthesis capabilities can support your long-term strategic goals in API development.