Scalable Metal-Free Synthesis of 3,4,5-Trisubstituted 1,2,4-Triazoles for Commercial API Production
Introduction to Advanced Triazole Synthesis Technology
The 1,2,4-triazole scaffold represents a cornerstone structural motif in modern medicinal chemistry, serving as a critical pharmacophore in a vast array of bioactive molecules. As illustrated in the structural diversity of approved pharmaceuticals, this heterocyclic system is integral to the efficacy of major drugs such as Maraviroc, Sitagliptin, and Deferasirox. 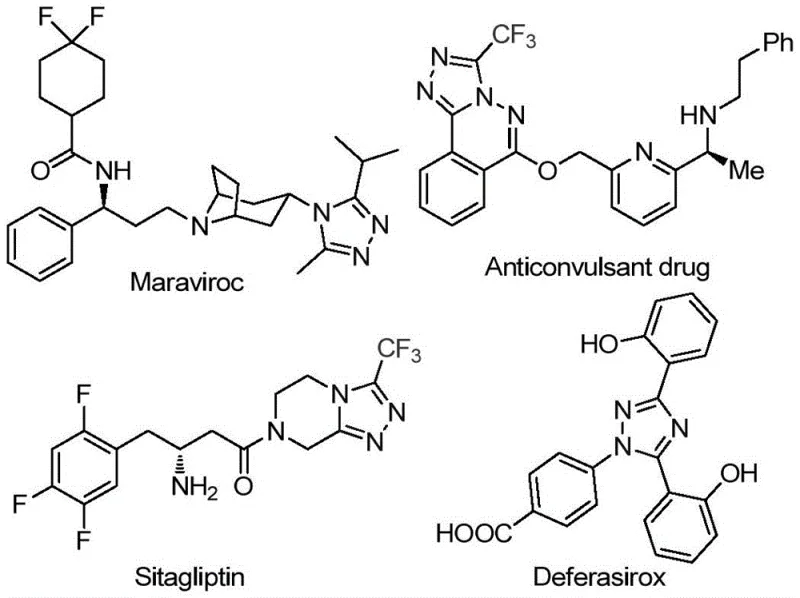 . The introduction of a trifluoromethyl group onto this scaffold further enhances metabolic stability and lipophilicity, properties that are highly sought after in lead optimization campaigns. However, the simultaneous installation of trifluoromethyl and acyl groups at the 3, 4, and 5 positions has historically presented significant synthetic challenges. Patent CN113105402B addresses this gap by disclosing a robust, metal-free preparation method that leverages inexpensive aryl ethanones and trifluoroethylimine hydrazides. This technological breakthrough offers a streamlined pathway for producing high-purity pharmaceutical intermediates, directly addressing the needs of R&D teams seeking efficient routes for complex heterocyclic libraries.
. The introduction of a trifluoromethyl group onto this scaffold further enhances metabolic stability and lipophilicity, properties that are highly sought after in lead optimization campaigns. However, the simultaneous installation of trifluoromethyl and acyl groups at the 3, 4, and 5 positions has historically presented significant synthetic challenges. Patent CN113105402B addresses this gap by disclosing a robust, metal-free preparation method that leverages inexpensive aryl ethanones and trifluoroethylimine hydrazides. This technological breakthrough offers a streamlined pathway for producing high-purity pharmaceutical intermediates, directly addressing the needs of R&D teams seeking efficient routes for complex heterocyclic libraries.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthetic routes for constructing polysubstituted 1,2,4-triazoles often rely heavily on transition metal catalysis or harsh reaction conditions that are incompatible with sensitive functional groups. Many existing protocols require stringent anhydrous and oxygen-free environments, necessitating specialized equipment and increasing operational complexity. Furthermore, the reliance on precious metal catalysts introduces significant downstream purification burdens, as removing trace metal residues to meet pharmaceutical standards can be both time-consuming and costly. These conventional methods frequently suffer from limited substrate scope, failing to accommodate diverse electronic and steric environments on the aromatic rings. Consequently, the development of versatile methodologies that can tolerate a wide range of substituents while maintaining high efficiency remains a critical unmet need in fine chemical manufacturing.
The Novel Approach
The methodology outlined in the patent represents a paradigm shift by utilizing elemental iodine as a dual-function promoter in dimethyl sulfoxide (DMSO). This approach eliminates the need for toxic heavy metals, thereby simplifying the impurity profile and reducing environmental impact. The reaction proceeds through a tandem sequence where aryl ethanones undergo iodination and Kornblum oxidation to generate reactive aryl diketones in situ. These intermediates then condense with trifluoroethylimine hydrazides to form the final triazole core. This strategy not only utilizes cheap and readily available starting materials but also operates under relatively mild thermal conditions without the need for inert atmosphere protection. The operational simplicity and broad functional group tolerance make this novel approach exceptionally suitable for the cost reduction in pharmaceutical intermediate manufacturing, allowing for the rapid generation of diverse analogues.
Mechanistic Insights into Iodine-Promoted Tandem Cyclization
The core of this synthetic innovation lies in the intricate interplay between iodine and DMSO, which facilitates a cascade of oxidative transformations. Initially, the aryl ethyl ketone reacts with molecular iodine in DMSO to undergo alpha-iodination followed by Kornblum oxidation, effectively converting the methyl ketone into a highly reactive 1,2-dicarbonyl species. This oxidative step is crucial as it activates the substrate for subsequent nucleophilic attack. Upon the addition of trifluoroethylimine hydrazide, a condensation reaction occurs to form a hydrazone intermediate. The presence of base, specifically sodium dihydrogen phosphate and pyridine, along with additional iodine, promotes the final intramolecular cyclization. 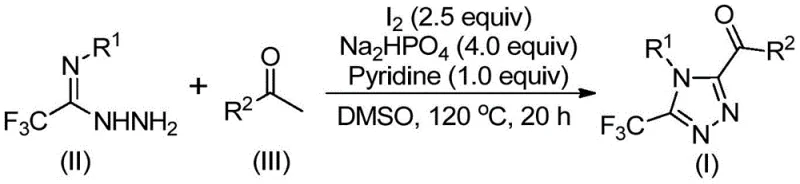 . This cyclization closes the five-membered ring, establishing the stable 1,2,4-triazole architecture with the trifluoromethyl group locked at the 3-position and the acyl group at the 5-position. The mechanism ensures high regioselectivity and minimizes the formation of isomeric byproducts, which is vital for maintaining high purity in the final API intermediate.
. This cyclization closes the five-membered ring, establishing the stable 1,2,4-triazole architecture with the trifluoromethyl group locked at the 3-position and the acyl group at the 5-position. The mechanism ensures high regioselectivity and minimizes the formation of isomeric byproducts, which is vital for maintaining high purity in the final API intermediate.
From an impurity control perspective, the absence of transition metals significantly reduces the risk of metal-catalyzed side reactions that often lead to complex impurity profiles. The use of DMSO as both solvent and oxidant participant ensures that the reaction medium remains homogeneous, facilitating heat transfer and mass transport. The specific stoichiometry of reagents, particularly the excess of aryl ethanone relative to the hydrazide, drives the equilibrium towards product formation while suppressing potential polymerization of the hydrazide. This mechanistic understanding allows process chemists to fine-tune reaction parameters, such as temperature and reaction time, to maximize yield and minimize waste, ensuring a robust process capable of delivering consistent quality across different batches.
How to Synthesize 3,4,5-Trisubstituted 1,2,4-Triazoles Efficiently
The practical implementation of this synthesis involves a straightforward one-pot procedure that can be adapted for both laboratory discovery and pilot plant operations. The process begins with the dissolution of aryl ethanone and iodine in DMSO, followed by a controlled heating phase to generate the dicarbonyl intermediate. Subsequent addition of the hydrazide and base components triggers the cyclization. Detailed standard operating procedures regarding exact stoichiometric ratios, specific temperature ramps, and workup protocols are essential for reproducibility. For a comprehensive guide on executing this transformation with optimal safety and efficiency, please refer to the standardized synthesis steps provided below.
- Mix aryl ethyl ketone and iodine in DMSO, heating to 90-110°C for 4-6 hours to initiate Kornblum oxidation.
- Add trifluoroethylimide hydrazide, sodium dihydrogen phosphate, pyridine, and additional iodine to the mixture.
- Heat the reaction to 110-130°C for 12-20 hours to complete cyclization, followed by filtration and column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this iodine-promoted synthesis offers tangible strategic benefits that extend beyond mere chemical novelty. The elimination of expensive palladium or copper catalysts translates directly into substantial cost savings on raw material procurement. Furthermore, the simplified purification process reduces the consumption of silica gel and solvents during downstream processing, lowering the overall cost of goods sold. The use of commodity chemicals like acetophenones and elemental iodine ensures a stable and resilient supply chain, mitigating the risks associated with sourcing specialized reagents. This reliability is crucial for maintaining continuous production schedules and meeting tight delivery deadlines for global pharmaceutical clients.
- Cost Reduction in Manufacturing: By replacing precious metal catalysts with inexpensive elemental iodine, the process drastically lowers the direct material costs associated with catalytic cycles. The avoidance of complex metal scavenging steps further reduces operational expenditures related to purification and waste disposal. Additionally, the high atom economy of the tandem reaction minimizes the generation of chemical waste, contributing to a more sustainable and economically viable manufacturing model. These factors collectively enhance the profit margins for contract manufacturing organizations and internal production units alike.
- Enhanced Supply Chain Reliability: The starting materials, including various substituted acetophenones and hydrazides, are widely available from multiple global suppliers, reducing dependency on single-source vendors. This diversification of the supply base enhances resilience against market fluctuations and logistical disruptions. The robustness of the reaction conditions, which do not require rigorous exclusion of moisture or oxygen, simplifies the engineering controls needed for production, allowing for faster turnaround times and more flexible scheduling. This operational flexibility is a key asset in managing the dynamic demands of the pharmaceutical market.
- Scalability and Environmental Compliance: The protocol has been demonstrated to scale effectively from gram-level experiments to larger batches without significant loss in efficiency, indicating strong potential for commercial scale-up of complex pharmaceutical intermediates. The use of DMSO, a solvent with a well-understood safety profile, and the absence of heavy metals simplify regulatory compliance and environmental permitting. The reduced toxicity profile of the reagents and byproducts aligns with green chemistry principles, facilitating easier waste treatment and disposal. This alignment with environmental standards is increasingly important for maintaining corporate social responsibility goals and meeting stringent regulatory requirements.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These answers are derived directly from the experimental data and beneficial effects reported in the patent documentation, providing clarity on the practical aspects of the method. Understanding these details is essential for evaluating the feasibility of integrating this route into existing manufacturing workflows.
Q: Does this synthesis require expensive heavy metal catalysts?
A: No, the method described in patent CN113105402B utilizes elemental iodine as a non-metal promoter, eliminating the need for costly transition metal catalysts and simplifying purification.
Q: What are the key advantages for large-scale manufacturing?
A: The process uses cheap, commercially available starting materials like aryl ethyl ketones, operates without strict anhydrous or oxygen-free conditions, and has been demonstrated to scale easily from gram to industrial levels.
Q: Can this method tolerate diverse functional groups?
A: Yes, the reaction exhibits broad substrate scope, successfully accommodating substituents such as methyl, methoxy, chloro, and trifluoromethyl groups on both the N-aryl and acyl moieties.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3,4,5-Trisubstituted 1,2,4-Triazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that advanced synthetic methodologies play in accelerating drug development timelines. Our team of expert process chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that promising laboratory discoveries can be seamlessly transitioned to industrial reality. We are committed to delivering high-purity 3,4,5-trisubstituted 1,2,4-triazole compounds that meet stringent purity specifications, supported by our rigorous QC labs and state-of-the-art analytical capabilities. Our dedication to quality assurance guarantees that every batch delivered adheres to the highest international standards, providing our partners with the confidence they need to advance their clinical programs.
We invite you to collaborate with us to leverage this innovative iodine-promoted synthesis for your next project. By partnering with NINGBO INNO PHARMCHEM, you gain access to a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality targets. We encourage you to contact our technical procurement team today to request specific COA data and route feasibility assessments. Let us help you optimize your supply chain and achieve your commercial objectives with our reliable 3,4,5-trisubstituted 1,2,4-triazole supplier services.