Optimizing Paxlovid Intermediate Production: A Convergent 3-Step Synthetic Strategy for Commercial Scale
Introduction to Advanced Paxlovid Intermediate Synthesis
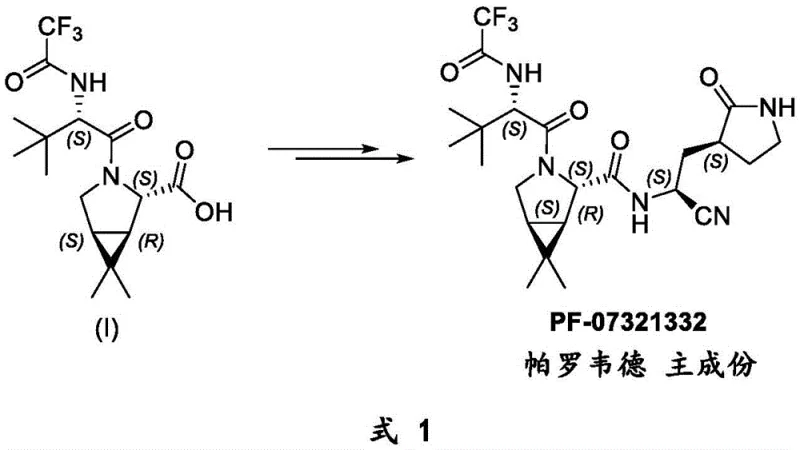
The global demand for effective antiviral therapeutics has placed immense pressure on the pharmaceutical supply chain to deliver high-purity active pharmaceutical ingredients (APIs) and their critical precursors with unprecedented speed and reliability. Patent CN114031543A introduces a transformative approach to synthesizing Compound I, a pivotal intermediate for PF-07321332, the main protease inhibitor component of Paxlovid. This innovation addresses the critical bottlenecks associated with traditional linear syntheses by implementing a highly efficient convergent strategy. By reducing the synthetic sequence from five steps to merely three, this methodology not only accelerates the time-to-market for bulk manufacturing but also significantly mitigates the accumulation of impurities that often plague longer synthetic routes. For R&D directors and process chemists, this represents a paradigm shift towards more sustainable and economically viable production of complex peptidomimetic structures.
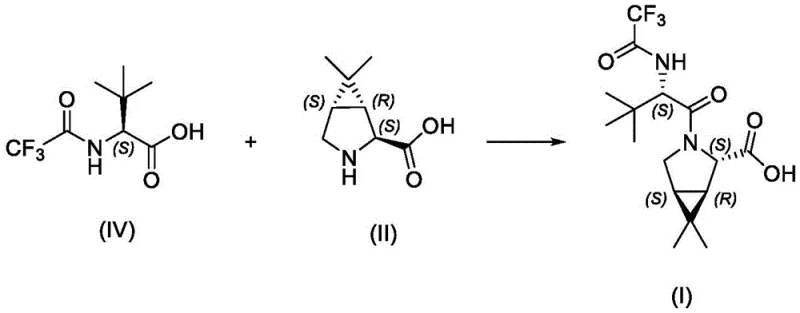
The core of this technological breakthrough lies in the strategic disconnection of the amide bond linking the cyclopropyl-fused pyrrolidine scaffold with the trifluoroacetyl-protected tert-leucine moiety. Unlike previous methods that relied on transient Boc-protection groups requiring harsh acidic deprotection conditions, this novel route utilizes a stable trifluoroacetyl group that serves both as a protecting group during synthesis and as a functional component of the final drug molecule. This dual functionality eliminates two entire synthetic operations—protection and deprotection—thereby streamlining the workflow. For procurement managers, this reduction in unit operations translates directly into lower operational expenditures (OpEx) and a reduced carbon footprint, aligning perfectly with modern green chemistry initiatives while ensuring a robust supply of reliable pharmaceutical intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of complex protease inhibitors like PF-07321332 has been hindered by inefficient linear pathways that suffer from low overall yields and high material costs. The original route disclosed in supplementary materials by Pfizer involves a sequential assembly where L-tert-leucine must first be protected with a Boc group, coupled to the proline scaffold, hydrolyzed, deprotected under acidic conditions to reveal the amine, and finally acylated with a trifluoroacetyl source. This five-step sequence is inherently flawed for large-scale manufacturing because each additional step introduces a multiplicative loss in yield and generates significant chemical waste. Furthermore, the use of strong acids for Boc-deprotection poses risks of epimerization at the chiral centers, potentially compromising the stereochemical integrity of the final API, which is a critical quality attribute that R&D teams must rigorously control.
The Novel Approach
In stark contrast, the method described in CN114031543A employs a convergent synthesis that dramatically simplifies the molecular construction. By preparing the N-trifluoroacetyl-L-tert-leucine fragment independently and coupling it directly with the hydrolyzed cyclopropyl proline acid, the process bypasses the need for orthogonal protecting group strategies entirely. This approach allows for the parallel optimization of two simpler sub-units, enhancing process robustness. The elimination of the Boc group manipulation not only shortens the timeline for cost reduction in pharmaceutical intermediates manufacturing but also removes the necessity for handling corrosive acids and the associated neutralization waste streams. This streamlined architecture ensures that the final product is obtained with higher purity profiles, reducing the burden on downstream purification processes such as chromatography or recrystallization.
Mechanistic Insights into Amide Bond Formation Strategies
The success of this convergent route hinges on the efficient activation of the carboxylic acid moiety of the N-trifluoroacetyl-L-tert-leucine (IV) to facilitate nucleophilic attack by the secondary amine of the cyclopropyl proline derivative. The patent elucidates several mechanistic pathways to achieve this transformation, providing flexibility for process optimization. One preferred embodiment involves the formation of a mixed anhydride intermediate using reagents such as p-toluenesulfonyl chloride or pivaloyl chloride in the presence of a tertiary amine base. This activation method generates a highly reactive electrophilic species that readily couples with the amine salt of the proline fragment. The mechanistic advantage here is the rapid reaction kinetics at mild temperatures, which minimizes the risk of racemization at the alpha-carbon of the amino acid, a common pitfall in peptide synthesis that can lead to difficult-to-remove diastereomeric impurities.
Alternatively, the patent describes the use of modern coupling reagents like T3P (n-propylphosphonic anhydride) or carbodiimides combined with additives like HOBT. These reagents operate through the formation of active esters or acyl phosphonates in situ, which are exceptionally efficient for sterically hindered couplings involving tert-leucine. The use of T3P is particularly noteworthy for commercial scale-up of complex pharmaceutical intermediates because its byproducts are water-soluble phosphonic acids, which can be easily removed during the aqueous workup phase. This simplifies the isolation procedure, avoiding the need for column chromatography and enabling the production of high-purity material through simple crystallization. Such mechanistic elegance ensures that the process remains scalable from kilogram to multi-ton quantities without sacrificing yield or quality.
How to Synthesize Paxlovid Intermediate Efficiently
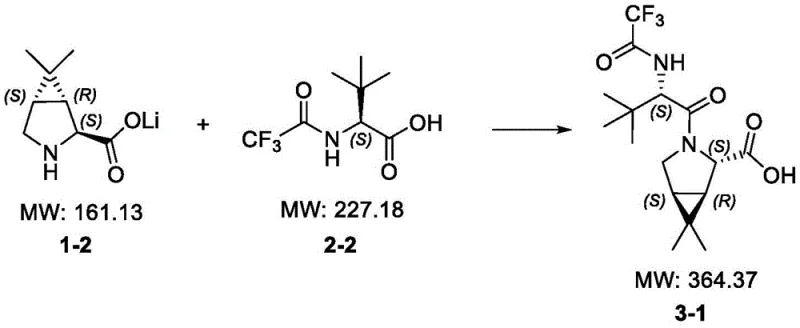
The execution of this synthesis requires precise control over reaction parameters to maximize the efficiency of the convergent coupling. The process begins with the independent preparation of the two key building blocks: the cyclopropyl-fused proline acid and the trifluoroacetyl-protected tert-leucine. The hydrolysis of the proline methyl ester is conducted under mild basic conditions using lithium hydroxide in tetrahydrofuran, ensuring complete conversion without ring-opening of the strained cyclopropane system. Simultaneously, the tert-leucine fragment is prepared by acylation with ethyl trifluoroacetate, a reaction that proceeds smoothly to provide the necessary activated acid component. These two streams are then merged in the final coupling stage, where the choice of activation method dictates the specific workup protocol. Detailed standardized synthesis steps for optimizing yield and purity are provided in the technical guide below.
- Hydrolyze methyl (1S,3aS,4aR)-4,4-dimethyl-2,3,3a,4a-tetrahydro-1H-cyclopropa[1,2-c]pyrrole-1-carboxylate (III) using inorganic bases like LiOH to obtain the free acid (II).
- Protect L-tert-leucine (V) by reacting with ethyl trifluoroacetate in the presence of a base to form N-trifluoroacetyl-L-tert-leucine (IV).
- Condense intermediates (II) and (IV) using coupling agents like T3P or via active ester formation (mixed anhydride/chloroformate) to yield the final target compound (I).
Commercial Advantages for Procurement and Supply Chain Teams
For supply chain leaders and procurement specialists, the adoption of this patented synthetic route offers compelling strategic advantages that extend beyond mere technical feasibility. The primary value driver is the substantial reduction in raw material consumption and processing time inherent in a three-step versus a five-step sequence. By eliminating the Boc-protection cycle, manufacturers can significantly reduce the volume of solvents and reagents required per kilogram of output, leading to drastic simplifications in waste management and disposal costs. This efficiency gain is critical in a high-volume market environment where margin compression is a constant challenge, allowing suppliers to offer more competitive pricing structures while maintaining healthy profitability. Furthermore, the use of commodity chemicals like L-tert-leucine and ethyl trifluoroacetate ensures a stable and diversified supply base, reducing the risk of shortages associated with exotic or proprietary reagents.
- Cost Reduction in Manufacturing: The elimination of two synthetic steps directly correlates to a decrease in labor, energy, and equipment usage. Without the need for separate protection and deprotection reactors, the capital expenditure required for facility setup is lowered, and the throughput of existing infrastructure is effectively doubled. Additionally, the avoidance of expensive coupling additives that generate difficult-to-remove urea byproducts (when using specific carbodiimides) in favor of cleaner activators like T3P or mixed anhydrides further drives down the cost of goods sold (COGS). This lean manufacturing approach ensures that the production of high-purity pharmaceutical intermediates remains economically sustainable even under fluctuating raw material price conditions.
- Enhanced Supply Chain Reliability: The convergent nature of this synthesis decouples the production of the two main fragments, allowing for parallel processing which acts as a buffer against supply disruptions. If there is a delay in the supply of one precursor, the other can still be manufactured and stocked, maintaining overall line readiness. Moreover, the robustness of the trifluoroacetyl group means the intermediate is stable and can be stored for extended periods without degradation, facilitating better inventory management and just-in-time delivery models. This stability is crucial for reducing lead time for high-purity pharmaceutical intermediates, ensuring that downstream API manufacturers receive materials exactly when needed without quality concerns.
- Scalability and Environmental Compliance: The process utilizes solvents such as tetrahydrofuran, isopropyl acetate, and n-hexane, which are well-understood in industrial settings and have established recovery and recycling protocols. The aqueous workup procedures described, particularly those involving T3P, generate benign byproducts that simplify wastewater treatment compliance. This alignment with environmental, social, and governance (ESG) goals makes the technology attractive for multinational corporations seeking to minimize their ecological footprint. The ability to scale from laboratory benchtop to multi-ton commercial production without fundamental changes to the chemistry ensures a seamless technology transfer, mitigating the risks typically associated with process scale-up.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis platform. These insights are derived directly from the experimental data and embodiments disclosed in the patent documentation, providing a factual basis for decision-making. Understanding these nuances is essential for technical teams evaluating the feasibility of adopting this route for their own manufacturing pipelines or for procurement officers assessing supplier capabilities.
Q: How does this new route improve upon the original Pfizer synthesis?
A: The original route disclosed in Science requires 5 steps involving Boc protection and deprotection cycles on L-tert-leucine. This patented method utilizes a convergent strategy that reduces the step count to just 3 reactions by directly using N-trifluoroacetyl-L-tert-leucine, eliminating the need for cumbersome protecting group manipulations and significantly improving overall atom economy.
Q: What coupling strategies are recommended for the final condensation step?
A: The patent discloses multiple viable options including mixed anhydride formation using p-toluenesulfonyl chloride, active ester formation using alkyl chloroformates, or direct coupling using modern reagents like T3P (n-propylphosphonic anhydride) or HATU. T3P offers a balance of high yield (up to 80%) and ease of workup due to water-soluble byproducts.
Q: Is this process suitable for large-scale manufacturing?
A: Yes, the process is designed for scalability. It avoids cryogenic conditions and uses common solvents like THF and isopropyl acetate. The convergent nature allows for the parallel production of the two key fragments (the cyclopropyl proline acid and the protected tert-leucine), which simplifies inventory management and reduces the risk of bottlenecks in commercial production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable PF-07321332 Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition from patent literature to commercial reality requires more than just chemical knowledge; it demands engineering excellence and rigorous quality assurance. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical benefits of this convergent synthesis are fully realized in practice. We have invested heavily in state-of-the-art reactor systems capable of handling the specific thermal and mixing requirements of amide couplings, alongside stringent purity specifications enforced by our rigorous QC labs. This infrastructure allows us to consistently deliver Compound I with the high stereochemical purity required for next-generation antiviral APIs, positioning us as a strategic partner for your long-term supply needs.
We invite you to engage with our technical procurement team to discuss how this optimized route can specifically benefit your project timelines and budget. By requesting a Customized Cost-Saving Analysis, you can gain a clear understanding of the economic impact of switching to this 3-step convergent method. We encourage potential partners to contact us for specific COA data and route feasibility assessments tailored to your volume requirements. Let us help you secure a resilient supply chain for this critical healthcare ingredient.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →