Scalable Synthesis of Anti-Inflammatory Ursolic Acid Derivatives for Commercial Production
Scalable Synthesis of Anti-Inflammatory Ursolic Acid Derivatives for Commercial Production
The pharmaceutical industry is constantly seeking novel scaffolds that offer improved therapeutic indices over natural products, and the modification of pentacyclic triterpenoids represents a frontier of significant interest. Patent CN102250188B discloses a robust and chemically elegant series of ursolic acid derivatives, specifically ursolic-2'-methoxy-4'(5')-[2-benzimidazole(benzothiazole)]-1'-phenyl esters, which exhibit potent anti-inflammatory and anti-tumor activities. This technology provides a critical pathway for transforming naturally occurring ursolic acid into high-value pharmaceutical intermediates through a sequence of esterification, heterocyclic cyclization, and selective deprotection. For R&D directors and procurement specialists, understanding the nuances of this synthetic route is essential for securing a reliable supply chain of these advanced bioactive compounds.
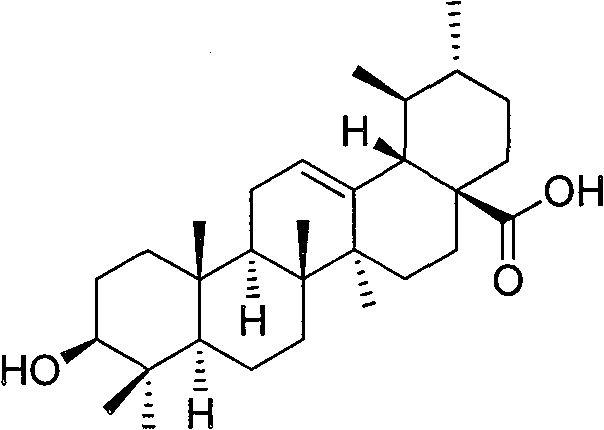
Ursolic acid itself is a widely distributed natural product, but its direct application is often limited by solubility issues and moderate potency. The innovation described in this patent addresses these limitations by introducing bulky heterocyclic moieties at the C-28 carboxylic acid position while simultaneously protecting and modifying the C-3 hydroxyl group. This dual-modification strategy not only enhances the lipophilicity and membrane permeability of the molecule but also introduces specific pharmacophores known for interacting with inflammatory pathways. The structural versatility allows for the generation of a library of analogs where R1 can be NH or S, and R2 can be H or OCH3, providing a diverse chemical space for drug discovery programs targeting chronic inflammation and oncology.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional methods for modifying the C-28 carboxylic acid of triterpenoids often suffer from harsh reaction conditions that can degrade the sensitive alkene functionality at the C-12 position or lead to unpredictable stereochemical outcomes at the C-3 position. Many conventional esterification protocols require strong acidic catalysts or coupling reagents that generate difficult-to-remove byproducts, complicating the downstream purification process and increasing the overall cost of goods. Furthermore, the introduction of complex heterocyclic rings like benzimidazoles or benzothiazoles onto the triterpene skeleton typically involves multi-step sequences with low atom economy, resulting in substantial waste generation and reduced overall yields. These inefficiencies pose significant challenges for supply chain heads who need to ensure consistent quality and volume for clinical trial materials.
The Novel Approach
The methodology outlined in patent CN102250188B offers a streamlined solution by utilizing trifluoroacetic anhydride (TFAA) as a mild activating agent for the initial esterification step. This approach allows for the formation of the phenyl ester linkage under relatively gentle conditions, preserving the integrity of the triterpene core. Subsequent cyclization reactions with o-phenylenediamine or o-aminothiophenol proceed efficiently in common solvents like ethanol or acetic acid, avoiding the need for exotic catalysts. The final deprotection step employs potassium bicarbonate in acetone, a weak base system that selectively removes the trifluoroacetyl group without affecting the newly formed heterocyclic rings or the ester linkage. This strategic design significantly simplifies the workflow, reduces the number of purification steps, and enhances the feasibility of large-scale manufacturing.
Mechanistic Insights into Esterification and Heterocyclic Cyclization
The core of this synthetic strategy lies in the sequential functionalization of the ursolic acid backbone, beginning with the activation of the C-28 carboxyl group. In the presence of trifluoroacetic anhydride, the carboxylic acid is converted into a mixed anhydride intermediate, which is highly reactive towards nucleophilic attack by the phenolic hydroxyl group of the substituted benzaldehyde. This reaction is typically conducted in toluene at elevated temperatures, facilitating the removal of trifluoroacetic acid byproduct and driving the equilibrium towards the desired ester. The resulting intermediate retains a trifluoroacetoxy group at the C-3 position, which serves as a temporary protecting group to prevent side reactions during the subsequent cyclization steps.
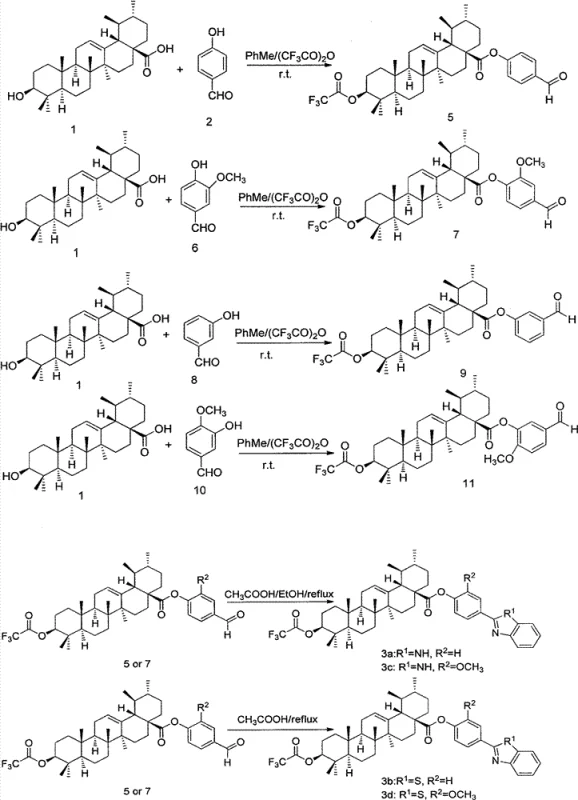
Following the esterification, the aldehyde moiety on the phenyl ring undergoes a condensation reaction with either o-phenylenediamine or o-aminothiophenol. In the case of benzimidazole formation, the reaction proceeds via a Schiff base intermediate which subsequently cyclizes and oxidizes to form the aromatic heterocycle. For benzothiazole derivatives, the mechanism involves a similar condensation followed by intramolecular cyclization driven by the nucleophilicity of the sulfur atom. These transformations are catalyzed by acetic acid or occur under thermal reflux conditions, ensuring high conversion rates. The final step involves the hydrolysis of the C-3 trifluoroacetoxy group using aqueous potassium bicarbonate in acetone. This mild basic condition is crucial as it cleaves the ester bond at C-3 to regenerate the free hydroxyl group while leaving the C-28 phenyl ester intact, demonstrating excellent chemoselectivity.
How to Synthesize Ursolic Acid Benzimidazole Esters Efficiently
The synthesis of these high-value intermediates requires precise control over reaction parameters to maximize yield and purity. The process begins with the dissolution of ursolic acid in toluene, followed by the addition of trifluoroacetic anhydride at room temperature to ensure complete activation before heating. The subsequent addition of the hydroxybenzaldehyde derivative must be carefully timed to minimize polymerization side reactions. For the cyclization step, the stoichiometry between the aldehyde intermediate and the diamine or aminothiol is critical, typically maintained at a 1:1 molar ratio to prevent excess reagent contamination. Detailed standardized synthesis steps see the guide below.
- Activate ursolic acid with trifluoroacetic anhydride in toluene and react with hydroxybenzaldehyde to form the trifluoroacetoxy ester intermediate.
- Perform condensation with o-phenylenediamine or o-aminothiophenol in ethanol or acetic acid under reflux to form the benzimidazole or benzothiazole ring.
- Deprotect the 3-position trifluoroacetoxy group using potassium bicarbonate in acetone to yield the final ursolic acid derivative.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement perspective, the adoption of this synthetic route offers substantial benefits regarding raw material availability and process safety. Ursolic acid is a naturally abundant compound extracted from various plant sources, ensuring a stable and cost-effective supply of the starting material. The reagents used, such as trifluoroacetic anhydride, hydroxybenzaldehydes, and o-phenylenediamine, are commodity chemicals available from multiple global suppliers, reducing the risk of supply chain bottlenecks. Furthermore, the solvents employed throughout the process—toluene, ethanol, acetic acid, and acetone—are standard industrial solvents with well-established recovery and recycling protocols, which contributes to significant cost reduction in pharmaceutical intermediate manufacturing.
- Cost Reduction in Manufacturing: The elimination of expensive transition metal catalysts and the use of simple workup procedures, such as filtration and crystallization, drastically lower the operational expenses associated with production. The high atom economy of the cyclization steps means less waste disposal cost, and the mild deprotection conditions reduce energy consumption compared to harsh acidic or basic hydrolysis methods. These factors combine to create a highly competitive cost structure for the final active pharmaceutical ingredients.
- Enhanced Supply Chain Reliability: Because the synthesis relies on robust chemical transformations that are insensitive to minor fluctuations in temperature or moisture, the process demonstrates high reproducibility across different batches and manufacturing sites. This reliability is paramount for maintaining continuous supply to downstream drug formulation teams. The use of common equipment and standard operating conditions further ensures that the technology can be easily transferred between contract manufacturing organizations without extensive re-validation.
- Scalability and Environmental Compliance: The process is inherently scalable, having been demonstrated effectively from laboratory scale to pilot scale without loss of efficiency. The absence of heavy metals and the use of recyclable solvents align with modern green chemistry principles, simplifying regulatory compliance and environmental permitting. This makes the technology particularly attractive for companies aiming to reduce their carbon footprint while scaling up complex pharmaceutical intermediates for commercial production.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these ursolic acid derivatives. The answers are derived directly from the experimental data and process descriptions found in the patent literature, providing a factual basis for decision-making.
Q: What are the key advantages of this synthesis route for ursolic acid derivatives?
A: The route utilizes mild activation conditions with trifluoroacetic anhydride and avoids heavy metal catalysts, ensuring a cleaner impurity profile and easier purification suitable for pharmaceutical applications.
Q: Can this process be scaled for industrial production?
A: Yes, the process uses common solvents like toluene, ethanol, and acetone, and operates at atmospheric pressure with standard reflux temperatures, making it highly amenable to commercial scale-up.
Q: What is the biological activity profile of these derivatives?
A: Specific derivatives, such as the 2'-methoxy-4'-(2-benzothiazole) and 2'-methoxy-5'-(2-benzimidazole) variants, have demonstrated significant anti-inflammatory activity in preclinical models.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Ursolic Acid Derivatives Supplier
At NINGBO INNO PHARMCHEM, we recognize the immense potential of triterpenoid-based therapeutics in the modern pharmaceutical landscape. Our team of expert chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project transitions smoothly from benchtop discovery to market-ready supply. We maintain stringent purity specifications and operate rigorous QC labs to guarantee that every batch of ursolic acid derivatives meets the highest international standards for safety and efficacy.
We invite you to contact our technical procurement team to discuss your specific requirements for these anti-inflammatory intermediates. By partnering with us, you gain access to a Customized Cost-Saving Analysis tailored to your volume needs, along with specific COA data and route feasibility assessments. Let us help you accelerate your drug development timeline with our reliable supply and technical expertise.